 "
"











Before we get started:
Biofilm formation is part of the reason S. mutans is so devastating to oral health (see [Project Overview] for more information). For adequate oral protection, biofilm formation is a problem we must solve, and we can do so internally, by modifying the S. mutans genome, (see [Control: Inhibitor]), or externally, via a modified probiotic. This means we need to know about conditions unique to biofilms in order to design a sensor.
What happens to our teeth if it is covered in biofilm? Metabolic acids which are waste products of S. mutans naturally begin to accumulate to very high concentrations in the local space, and cannot disperse out into the rest of the mouth. This results in a low pH in areas covered by biofilms, about 4.0~5.0. Since our probiotic is physically close to S. mutans (see [Cleanse: Attachment]), if our probiotic detects a low pH, there’s a good chance it is within a biofilm. Figure 1 depicts this in a simplified diagram.

Figure 1.
Now that we know the target pH range, we need to find out what biofilms are made of, and how to get rid of them. Biofilms are actually conglomerated bacteria colonies enmeshed in a self-synthesized layer of extracellular polysaccharide matrix. Of these, PGA, or poly-gamma glutamate, has been demonstrated to be a main component across the biofilm of several phylogenetically diverse bacterial species, and polyglycine bridges can cross-link glycopeptides that make up the cell walls of biofilm-producing bacteria.
The ideal biofilm degrader must be able to target some, if not all, of these elements.
So how did we do it?
After careful literature search, we found two promising enzymes:
Lysostaphin is a zinc-containing enzyme 27kDa in size, taken from the genome of Staphylococcus simulans, and was used previously by the [2012 HIT-Harbin iGEM team]. It is particularly powerful because it combines the actions of three different peptidoglycan degradation enzymes, and as such can target a wide variety of the glycopeptides that form cell walls, including features such as the polyglycine bridge. Also, unlike other enzymes of this type, it can target both actively growing and non-dividing cells. To add to its merits, lysostaphin causes no side effects in humans.
Dispersin B comes from Aggregatibacter actinomycetemcomitans, a Gram-negative rod oral bacteria. It is a soluble glycoside hydrolase capable of degrading PGA.
After the enzymes have been decided, we needed to find a promoter responsive to the conditions within a biofilm. The Asr promoter comes from E. coli strain MG1655, and is induced by extracellular low pH of between 4.0~5.0. Perfect for what we need.
But wait! We’re not quite done yet. Even after being equipped with the Asr promoter, lysostaphin, and dispersin B, our probiotic would still be useless against biofilms unless we export the enzymes out of the cell, where they can do their jobs. Here’s where the signal protein comes in.
YebF is a signal protein naturally secreted by E. coli, though experimental strains rarely secrete this protein, which makes it useful in synthetic biology. Proteins tacked onto YebF can be expected to also be secreted outside of the host cell, so we put this in front of our enzymes.
At last, our circuit is complete!

Our expectation is that the three-protein fusion of YebF-Lysostaphin-Dispersin B will be exported out of the cell upon translation by the YebF, where they will be able to do their job of degrading the biofilm, such as in Figure 2.
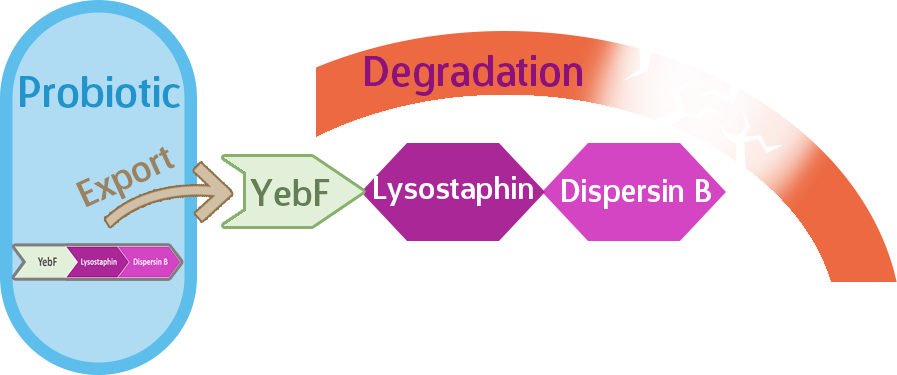
Figure 2.
Putting it to the test!
Having a circuit is only the start! Now we must test how well our circuit functions. For the Antibiofilm circuit, we need to test specifically for three things: the function of the Asr promoter, the function of YebF, and the function of lysostaphin and dispersin B.
Testing the Function of the Asr Promoter
For this test we constructed the following circuit in E. coli. We aim to elucidate the exact pH-activity relation by subjecting the circuit to conditions of various pH, and measuring activity via RFP fluorescence intensity, quantized in OD.

Here is the details of our method:
- Transform the circuit to E. coli, and grow the modified E.coli on agar plates overnight.
- Select the strain that expresses growth differences from the previous step. Prepare two of each liquid culture tubes for pH values 3.5, 4, 4.5, 5, 5.5, 6, 6.5, and 7, and grow the selected strain in all tubes. Use the growth data from each tube to plot the precise effect of pH on the Asr promoter.
- Measure the OD value (RFP/bacterium number), and plot out curves.
Testing the Function of YebF
Since we also used YebF in [Control: Target], we have done our functional assays there. Feel free to check it out if you’re interested!
Testing the Function of Lysostaphin and Dispersin B
For this part of the test, we designed a novel apparatus we called the [HOPErfusion], which allowed us to culture S. mutans in real saliva (yes, real saliva!) and observe their biofilm formation. The HOPErfusion provided us with the most accurate representation of the human mouth that we could obtain!
Experimental Design
We have designed two circuits to treat the biofilm formed by S. mutans.


We cultured S. mutans overnight for 12 hours, and move cultured S. mutans to the modified slide glass which was coated by saliva(37°C, 1 hr). Then we used peristaltic Pump to drop BHI and saliva on slide glass to simulate the flowing atmosphere of oral cavity. All the experiments are conducted in our designed device, which is closed atmosphere mimicking the anoxic atmosphere.
To test whether our modified E. coli has the ability in decreasing formed biofilm, we put modified E. coli (circuit 1 and 2) to treat S. mutans for 12 hr, 24hr and 36hr. Finally, we did biofilm grow assay via crystal violet stain. biofilm grow assay:
- gentally washing with ddH2O three times.
- Air dried at room temperature
- Stain with 500ul 0.1% crystal violet for 15min at room temperature
- Add ddH2O three times to wash excess crystal violet
- Add 1000 atic acid to extract crystal violet stain and wait for10 min.
- Add 20 extract crystal violet stain and 800 ddH2O to do OD measurement at 595nm wavelength.

AIR DRIVED AT ROOM TEMPERATURE

ADD ddH2O THREE TIMES TO
WASH EXCESS CRYSTAL VIOLET
Our result
experiment1: using Hoperfusion to do biofilm growth assay
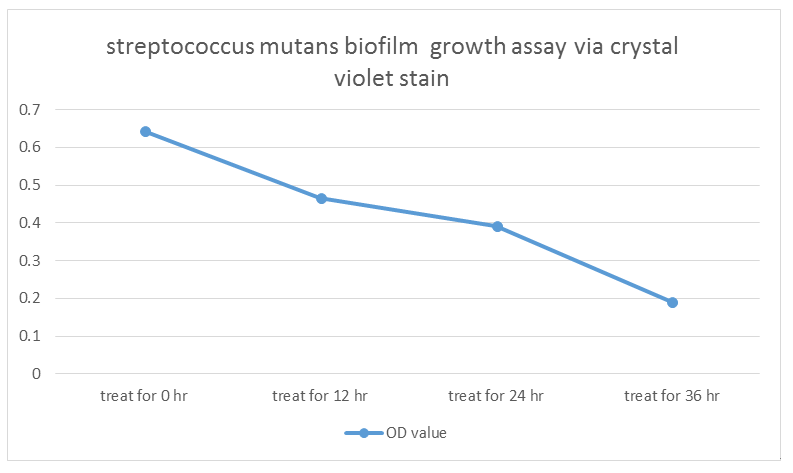
- S. mutans growing for 36 hr (control) OD = 0.643
- S. mutans growing for 36 hr and treat with our modified E. coli for 12hr OD =0.465
- S. mutans growing for 36 hr and treat with our modified E. coli for 24hr OD =0.391
- S. mutans growing for 36 hr and treat with our modified E. coli for 36hr OD =0.191
Indicated from the graph, we could clearly see that treating our modified E. coli (transformed by Dispersin B circuit and lysostephin circuit ) have a negative impact on biofilm growth of Streptococcus mutans. As a result, we have confidence that our modified E. coli have ability to treat or influence biofilm formation.


This is the picture that We co-cultured S. mutans and our modified E. coli through different time span on slide glass coated by saliva and do biofilm grow assay via crystal violet stain and test OD.
(1)S. mutans growing for 36 hr (control) OD = 0.643
(2)S. mutans growing for 36 hr and treat with our modified E. coli for 12hr OD =0.465
(3)S. mutans growing for 36 hr and treat with our modified E. coli for 24hr OD =0.391
(4)S. mutans growing for 36 hr and treat with our modified E. coli for 36hr OD =0.191









