 "
"
Team:Calgary/Notebook/Journal/Detectives
From 2014.igem.org

Detectives' Journal
Week 1: May 12 - 16
This week we spent most of our time doing a literature search on potential systems to recognize DNA sequences. One potential system included homologous recombination - homologous sequences being a) the PCR-amplified target pathogen DNA and b) a specific region of genomic (or plasmid) DNA; homologous recombination would activate a genetic circuit leading to expression of a reporter. The other potential system would be via cis-repressed mRNA - mRNA would become ‘readable’ (i.e. repression would be released) upon hybridization with the complementary target pathogen DNA sequence. We also looked at potential promoters for B. subtilis that are repressible, as well as potential reporters, such as chromoproteins and enzymes which produce coloured products, to use in our reporter system.
We were also able to clone several iGEM registry parts (RFP, eCFP, GFP, YFP, pTetR, pLacI, RBS, RFP-generator, and a ‘double terminator’). RFP, eCFP, GFP and YFP were cloned to be used as reporters. The pTetR and pLacI were cloned to be later used as binding sites for our reporter system.
Week 2: May 19 - 23
Again, we spent more time on literature research, specifically in regards to the proposed detection strategies; mRNA cis-repression and homologous recombination. We read more about the detailed mechanisms involved in homologous recombination. We learned that there is overlap between the RecA mechanism of natural transformation and the RecBCD mechanism during homologous recombination (Dillingham & Kowalczykowsk, 2008). Another endogenous homologous recombination system that could be used is via the RecF mechanism. If all else fails we could resort to the Lambda Red strategy. The advantage of using RecBCD would be in using ssDNA which is produced by the RecA system during transformation, thus RecA + RecBCD would be the preferred mechanism for homologous recombination.
Reading about cis-repression we learned that our main objective will be to design a suitable cis-repressor (crRNA) sequence. The crRNA must be complementary to both the RBS and the target DNA, with a greater affinity to the target DNA to promote reliable de-repression. The crRNA sequence does not have to necessarily cover the entire RBS sequence - only enough to inhibit the binding of the ribosome. According to literature (Isaacs et al 2004) the crRNA must bind to only a few of the nucleotides in the RBS sequence in addition to binding the 5’ UTR. Overall the crRNA sequence must be 19-22 nucleotides. The secondary structure is also important to consider - some bulges (i.e. mismatches) must be present in addition to an adequately sized loop to allow for reliable binding of the trans-activation sequence (the amplified target pathogen DNA sequence), otherwise the mRNA will be strongly repressed. Programs such as Mfold may be necessary to predict the secondary structure and thermodynamics of the system (Isaacs et al 2004).
We cloned several more parts; PlepA, Pveg, amilCP, LacI (+LVA), tetR (+LVA). The promoters that we cloned were PlepA and Pveg which are endogenous to B. subtilis, as well as a constitutive promoter. AmilCP is a blue chromoprotein that does not fluoresce and was cloned so that it could potentially be used in our reporter system. The repressors we cloned contain a rapid degradation tag (LVA) that will increase the proteins turnover rate and provide us with a faster response.
Week 3: May 26 - 30
We introduced ourselves to the online software ‘Benchling’ and began cataloging our parts. We will be using Benchling to keep track of our parts during the cloning process. We also used Benchling to construct our first reporter/repressor test systems in silico. Performing cloning in silico helped us get an idea of the steps that we will need to take, and to plan out the restriction digestions that we will need, i.e. how to use the BioBrick system to assemble our constructs and exactly which enzymes to use.
We have gently settled on the idea of using a two-part genetic circuit, where the repressor gene regulates the reporter gene in a fashion analagous to ‘gates’ in electrical engineering (Brophy & Voigt 2014). We cloned several more parts, specifically the repressor/promoter pairs: C1-lambda, C1-434, and C2-P22. Promoters will be used as operators - though they function as promoters, we will be using the strong, constitutive promoter Pveg instead, then the repressible promoters will instead function as an operator. By using a strong, constitutive promoter we will ensure optimal expression regardless of the operator used. We also repeated efforts to clone RBS, pTetR, and TetR - up to the point of completing miniprep and confirmation by colony PCR.
We requested more B. subtilis parts from iGEM HQ - a terminator, and several RBS sequences and plasmid vectors. All but one of the vectors will be an integration vector which will allow us to integrate our constructs into a specific locus in the B. subtilis genome. The RBS sequences will offer improved translation rates in B. subtilis compared to using RBS sequences originating from E. coli.
Week 4: June 2 - 6
This week we contacted both Dr. Lisa Allen and Dr. Abebe Bayih in hopes that they would be able to give insight on our multi-disease detector for a quick and easy diagnosis of neglected diseases in the developing world. Given that they have hands-on experience in impoverished areas, they are able to offer valuable opinions of what technology local people currently use and also what they would want created and/or improved. We also brainstormed further questions to ask. As a group we did a few trials of DNA extraction from strawberries in order to become proficient in our techniques for the Telus Spark adults only night the following week. We experimented with various strengths and types of alcohol, freezing, thawing, and incubation times and also whether or not the strawberries were frozen in order to optimize the process of making the “DNA-daiquiri’s”.
We continued our efforts to clone the parts necessary for our genetic circuit. Many of the small parts (e.g. promoters and RBS) could not be validated with confidence either with colony PCR or restriction digest because their sequences are so short, therefore we instead ordered PCR primers to ‘synthesize’ the small parts by essentially filling in the gaps at the ends. We also went over the BioBrick assembly scheme and are ready to begin assembly of our reporter/repressor genes.
We are still looking for a suitable reporter. At this point we think that the most appropriate reporter will likely be an enzyme capable of producing a colored product, this would allow for a large degree of signalling without the side effects of cytotoxicity from the accumulation of recombinatory protein. The enzyme could potentially produce more product and therefore more response than the production of recombinatory protein would allow. Two examples of potential enzymatic reporters found in the registry are the lacZ and xylE genes. We are also considering using a novel enzyme which we could characterize and submit to the iGEM registry.
Week 5: June 9 - 11
Assembly of our reporter/repressor constructs is predicted to be troublesome. To hedge our bets we began designing sequences for synthesis. These sequences are essentially identical to what our in-lab assembled constructs will be, including scars between the BioBricks (Fig. 1). Literature research was performed on the subject of reporters. We are interested in finding and using a novel reporter in the form of an enzyme/substrate, we feel that the enzyme/substrate may be the best choice since we can obtain a continuous signal without risking recombinant-protein toxicity. However, we are also exploring chromophoric proteins. We are considering the lux and electrochemical reporters for a potential ‘premium tier’ version of our detector which may be of use in more advanced laboratory settings.

Figure 1: Examples of the composite regulatory parts that we designed. Arrows represent the constitutive promoter Pveg; orange rectangles represent the repressible promoter, also referred to as the operator; blue and green semicircles represent the ‘weak’ or ‘consensus’ variants a RBS B. subtilis. Structures i and ii contain a single operator, whereas structures iii and iv contain a triplicate of operators adjacent to each other. The triplicate repeats of the operator were designed to allow stronger and more reliable repression, in an effort to reduce leakiness of expression. In the interest of characterizing and developing an optimal system, a total of five different operators were chosen, for a total of 20 unique composite regulatory parts.
We continued our efforts to validate the presence of our parts that we have miniprepped. Additionally, we “synthesized” some of the small parts such as promoters, notably pTetR due to the several failed attempts of isolating it. We also received several plasmid vectors for use in B. subtilis from iGEM HQ, which were streaked over the weekend. Unfortunately we had some troubles with contamination, as such we are still trying to obtain the necessary parts.
We spent a few days preparing for our first outreach program at Calgary’s Science Centre. The topic was Food Science. We practiced (and enjoyed) a protocol for the extraction of DNA from strawberries, which ultimately resulted in strawberry daiquiris. We also explained to the public the molecular mechanisms behind certain food-related phenomena such as why banana’s go brown, and why browned meat tastes better as well as the flavor of unami.
Week 6: June 16 - 20
Our ‘synthesized’ parts - pTetR, pC1-lambda, pC1-434, pC2-P22, RBS x2 - were ligated into plasmid vectors, and subsequently validated. We now have all of the parts necessary for the assembly of our constructs. Additionally we prepared constructs for synthesis, a total of 20 distinct reporter genes. The genes will vary with respect to 2 different RBSs, and 5 different operators (repressible promoters). Additionally, we created one set with a single operator, and another set with 3x of each promoter directly adjacent. We would like to be sure that our reporter is tightly regulated, eventually we will perform a quantitative analysis of each.
After further investigation into literature and discussion, the crRNA strategy may not be as viable as initially thought. The upstream ‘cr’ sequence must be complementary to the entire RBS, which is a problem because it must also be complementary to the target DNA sequence - a sequence which we have very little control over. According to the literature, there are multiple parameters in regards to the sequence design which must be considered, including optimization of the dynamic range - tight regulation with a high level of expression. Furthermore, the post-transcriptional nature of this strategy may be more susceptible to leaky expression, which for our purpose, is highly undesirable.
Integration vector plasmids were also prepared, and ready for transformation of B. subtilis.
Week 7: June 23 - 27
We continued our efforts in building the repressor construct, consisting of Pveg + RBS [for E. coli] and one of five coding sequences for a repressor protein. A similar repressor genewill also be made consisting of two variants of an RBS - a RBS consensus sequence for B. subtilis and an RBS that works in both E. coli and B. subtilis.
The two RBS’s that will be used in B. subtilis were synthesized, because they were not immediately available from the iGEM kit plates. This week they were inserted into a plasmid, after several attempts.
We have also been dealing with logistical problems in ordering the synthesis of our reporter construct ‘skeleton’ - that is the Pveg + [operator] + [RBS] - that will be used to make reporter genes with different reporters.
Week 8: June 30 - July 4
Attempted once more to clone the three different synthesized RBSs into a plasmid. Since the parts are so small they are difficult to verify, however, it appears that they were successfully cloned into a plasmid vector and into E. coli. Subsequently the constitutive promoter, Pveg, was inserted in front of the RBS.
Continuing effort in making the repressor/generator gene using a constitutive promoter and one of 3 RBSs - Pveg + RBS (x3) + [repressor or reporter]
Week 9: July 7 - 11
While waiting for synthesis of our reporter genes, we are looking for a similar gene- promoter + operator + RBS - to amplify out to use for testing our system, preferably from B. subtilis. However, since we do not want to have our system overlap with the native regulatory systems in B. subtilis, we will again attempt to assemble the reporter geneusing the parts that we have.
Continuing efforts to make our own ‘generators’ (e.g. RFP_gen) that is relevant to our project - using the relevant promoters and RBSs. This will provide us with other options if RFP is deemed suboptimal.
We have found an isothermal PCR (RPA) kit for use with our project. RPA was chosen as the best candidate because of it’s reasonably low operating temperature at 37-39C, compared to another commercially available isothermal PCR system, SDA, which has an optimal temperature 60+C.
We also had our second AITF GeekStarter workshop. A significant takeaway was learning about an alternative assembly method. Using the type IIs restriction enzyme BsaI, and custom primers that would allow us to produce custom sticky ends we could theoretically assemble our entire construct (4-6 parts) in one reaction, which would drastically cut down our time spent in the lab. This will be attempted in parallel to the traditional BioBrick assembly procedure.
Week 10: July 14 - 25
We have obtained a Blood PCR Kit from KAPA that allows direct PCR from whole blood without the isolation of DNA beforehand. We are also in the process of ordering an isothermal PCR kit (RPA; Recombinase Polymerase Amplification) from TwistDx. Since we intend on performing isothermal PCR on whole blood samples we are looking into the molecular details of the conditions in both KAPA Blood PCR and TwistDx basic RPA kit in order to effectively combine both technologies into a suitable strategy for us to obtain a prepared and PCR-amplified sample. We have asked for further support in the form of information from KAPA.
Primers that will allow us to carry out the modified method of ‘Golden Gate’ assembly have been placed for order, we are now waiting for them so that we may begin 1-step assembly. We are also waiting on our synthesized genes.
We have been contacted by 2 more groups so far for collaboration, details are being worked out. We now have three potential collaborations to work on from Purdue, Cornell and Colombia iGEM teams.
Efforts are still being made to assemble our genetic detection circuit manually, using the traditional BioBrick assembly method. However, many failures have been witnessed and it appears to be an uphill battle.
Week 11: Aug 4 - 8
At this time a few members from our group were at the Biological Weapons Convention in Geneva. We designed new primers for the target meningitis site, and for the homologous flanking sequences. One large sequence (3kb) used as our mock target DNA, and two smaller flanking regions (1kb each) that will be ligated to the repressor operon. LacZ and the Pxyl+comK sequences are being BioBricked. The lacZ currently in the registry (found here) is of poor quality and not available beyond the 2012 iGEM distribution plates. Pxyl+comK will allow for other teams to create super-competent B. subtilis cells. Although technically it isn’t increased competency, rather, it is competency that is induced by xylose.
Week 12: Aug 11 - 15
Most synthesized sequences have arrived, preparing to ligate reporter coding sequences to the synthesized genes and use them with the standard BioBrick vector plasmids. Since we have had a lot of trouble ligating the small RBS parts, we will be using the reporter genes as the genes for the repressors - all that is necessary is a constitutive promoter, we only need to make sure that there will be no overlap with the repressor/operators innately in B. subtilis. The promoter/operator/repressors C1-434, C2-P22, and C1-lambda originate from bacteriophages and so we do not expect activity in B. subtilis. Conversely, lacI is a common gene and according to our searches it exists in both B. subtilis and E. coli
Week 13: Aug 18 - 22
We continued working on assembling our reporter with the synthesized promoter/regulatory constructs. RFP is being used for now because it is readily available and familiar, but lacZ will be used as soon as it is available in BioBrick form. We are also planning on ligating the repressors to the synthesized constructs - we have not been able to assemble the repressor genes manually due to poor success with cloning the RBS.
We also worked on our collaboration with Colombia - developing and testing a “homebrew” method of making competent cells using reagents and equipment available outside of a laboratory. We will first attempt to use tap, bottled, and distilled water with calcium added via over-the-counter supplements.
The isothermal PCR kit from TwistDx has arrived. We are now preparing to i) individually test the isoPCR kit and the Blood PCR kit for efficacy with our mock pathogen, under standard conditions (i.e. isoPCR without blood ) and ii) test a hybrid PCR setup that utilizes both RPA (isoPCR) and the KAPA blood-compatible Taq in conjunction. Our experiments will use a mock-pathogen-contaminated blood sample - a mixture of blood and a liquid E. coli culture containing a known plasmid and gene.
Week 14: Aug 25-29
We have had little success with the synthesized genes in regards to cloning. After multiple attempts it was discovered that the plasmid containing our synthesized gene was not successfully transformed. This was indicated by running digested and undigested plasmid minipreps on a gel, which showed no bands. We are starting from scratch again - transforming E. coli with our synthesized product to obtain viable genes to clone. The construct that was previously apparently successfully assembled was sent for sequencing to verify.
Isothermal PCR appears to work. The positive control showed a smear caused by protein-bound DNA. Cleaning the PCR product with a purification column showed a sharp band at the expected size of 132bp. This was replicated by diluting the PCR products by at least a factor of 10, demonstrating that purification is not strictly necessary. The dilution of the PCR products promotes a dissociation of the protein and bound DNA. When using BioBrick primers to amplify a sequence many small bands <500bp were produced, this was expected as described by the TwistDx manual that optimal amplification occurs for amplicons 200-400bp. The TwistDx resources go on to describe the small bands as ‘primer noise’ that is inherent to the system. Reduction of primer noise is possible namely by screening several variants of primers. We are attempting to reduce the primer noise by synthesizing longer primers 34bp for improved specificity as recommended by TwistDx. Isothermal PCR appeared to work nearly equally well with both whole colonies and purified plasmid as the template (Fig. 2).

Figure 2: Results of isoPCR under standard isothermal conditions using BioBrick primers. Two different sequences were targeted for amplification; C1-λ is a promoter sequence with an expected amplicon size of 406bp; RFP is a coding sequence with an expected amplicon size of 1100bp. Templates used in this experiment were either whole colonies labelled ‘C’, or purified plasmid extracts labelled ‘P’.
Sheep blood treated with citrate (as an anticoagulant) was ordered and will arrive next week for further testing of isothermal PCR kit in the presence of blood.
Week 15: Sept 1 - 5
We restarted efforts to ligate a reporter or repressor to our synthesized construct. We also transformed E. coli with the synthesized parts, this time done properly with IPTG and X-gal on the agar plates - only few blue colonies were shown indicating that transformation was highly successful. Colonies were selected, used to make overnight cultures, from which plasmid DNA was isolated via the plasmid miniprep protocol. The C2-P22 repressor was ligated with the Pveg_C1-lambda_consensusRBS construct, using both single-operator and triple-operator variants. Likewise the RFP reporter was ligated with the Pveg_C2-P22_consensusRBS construct with both single- and triple-operator variants. The RFP-ligation allows for straightforward visual screening as successful ligations are indicated by red colonies. The repressor-ligations are being screened via colony-PCR.
We used the isothermal PCR kit for the first time and discovered a new problem - the PCR product contains protein-bound DNA which prevents visualization on agarose gels, and may pose a problem for transformation if DNA is not free. DNA purification is required, however, fortunately dissociation of protein-bound DNA may be induced via 1/10 dilution of the PCR product. This is promising as we would expect a similar dilution in our final prototype, we expect that the strong association between DNA and protein will not pose a problem.
The TwistDx RPA isothermal PCR kit was originally designed for rapidly amplifying short (<400bp) amplicons, unfortunately for our purposes we require amplification products to be closer to 1kbp in size. According to literature (Piepenburg et al. 2006) it is possible to amplify sequences of up to 1.5kbp using RPA. We are attempting to optimize conditions so that we obtain longer amplicons. We may adjust parameters such as the concentration of primers, magnesium-acetate, dNTP, and ATP, as well as incubation temperature - with the aim of increasing the elongation time while providing enough resources (i.e. ATP) to allow for a longer reaction time.
This week we spent a night at an outreach event "Beakerhead" downtown where we helped the public draw with fluorescent bacteria. We spent the weekend with the team at aGEM, where we gave a presentation of our project and received helpful feedback on it. During the meeting on Sunday, we took on the task of biobricking our 20 synthesized constructs so that they can be submitted to the registry. Below is a table that we made which we will update at the end of each coming week so that we can keep track of what parts get to what step, which ones succeed and also when some fail (which we all know they will) that we will know where they failed and then can begin taking it back a step (or two) to see if we can get it to work on a second try. We have given each part in the "synthesized construct" column a shortened name: easy to identify the full name if you go to the Benchling account and look under "Assembled Constructs". Some synthesized parts have not been able to be successfully transformed yet into E.coli. I am going to start with the ones that have and once I am through the process once I will go back and attempt to transform them again.
Week 16: Sept 8 - 12
Blood PCR has shown to work wonderfully, with PCR products formed at a blood concentration of 20% v/v; the KAPA blood PCR kit used claimed to have optimal efficiency up to 10% blood v/v. The blood-compatibility is conferred by a second-generation Taq polymerase, engineered via directed-evolution, that is resistant to polymerase inhibitors typically found in blood. For comparison, PCR with typical first-generation Taq showed drastically reduced efficiency of amplification at blood concentration of 1% v/v.
The next step was to show that isothermal PCR (RPA), supplemented with the blood-compatible Taq polymerase, would work efficiently even in the presence of blood. 25, 50, or 75% of the volume of the isothermal PCR reaction (i.e. the buffer and water) was substituted with the KAPA blood PCR mix. Results showed that indeed the blood PCR kit supplement allowed isothermal PCR to work in the presence of up to 10% v/v of blood, when 25% of the volume was by the blood PCR mix. Unfortunately at 50% and 75% substitution, the reaction did not work at all, this is most likely due to a lack of ATP - the isothermal PCR buffer contains ATP necessary for recombinase activity, at higher substitutions of blood PCR mix the buffer was not used or was not enough in regards to the amount of ATP present.
We are also working on determining the sensitivity of our amplification. Results will be judged by visualization on agarose gel, if no other means of detection of DNA amplicons are available.
Week 17: Sept 15 - 19
This week we began the process of preparing our synthesized genes for submission to the iGEM registry. We started with the 15 previously successful transformed parts and made overnights of two colonies from each construct. We did this twice for the parts that failed on the first try, with the second time using different colonies than the first. I mini-prepped all successful overnight cultures. There were 3 parts that failed growing in overnight cultures twice so we will come back to those later and try again. With the remaining 12 successful constructs (refer to column 3 or 4 of Table 1), we did a restriction digest. On Monday we will run these samples on a 1% gel and then cut out the correct band, either the part we want (insert) or the vector. This week we also tested the competent E.coli that we made last week and by sent lacZ for sequencing.
Week 18: Sept 22 - 26
This week we continued using the parts we digested last week. We started by doing a gel extraction of the digested samples. We then did a ligation of each sample insert with each of two vectors. We did colony PCR to verify that each colony contained the ligated product that was previously made. The first time we did this only 6 out of 66 colonies that we chose contained the ligated product. We tried a second attempt and another 6 colonies appeared to be successful. We sent 6/12 samples for sequencing so far. Next week we'll be sending the remaining 6 samples for sequencing. So far there are 6 parts total that have been mini-prepped to the step 9. Once the sequencing results are back next week we will have confirmation that these BioBricks are correct and ready to be submitted to the iGEM registry. Next week in addition to sending the remaining 6 samples for sequencing, we will be trying the process all over again to get as many more synthesized genes BioBricked as possible.
Week 19: Sept 29 - Oct 3
This week when we got the sequencing results back we discovered that the parts we thought had successfully been BioBricked were actually CjBlue, a chromoprotein, and not our synthesized constructs in the iGEM plasmid. We went back and realigned the sequences from other repressor ligations last week and it turns out that those samples were also CjBlue. We then went back and re-aligned the lacZ sequences that had been previously sent for sequencing and found that they too contained CjBlue. Thus, the mystery of the CjBlue began…
We started the process again by transforming more of our synthesized constructs into E. coli and mini-prepped those samples. To try to narrow down where CjBlue contamination was coming from we sent in all undigested mini-prepped samples of our synthesized constructs from this week and last week for sequencing. We also sent in our stock of mini-prepped vectors (used for plasmid switch our synthesized parts into) for sequencing to verify that these are RFP. and not the source of cj blue contamination. Once these results are back we will be one step closer to figuring out where the cj blue entered the picture. Our summer iGEM project was also presented at the University of Calgary BHSc research symposium. Finally, on Saturday, we went back to the cPCR agarose gels we ran last week, in which we chose colonies that we thought contained the correct construct in the iGEM plasmid. Looking back now it appears that these bands are a bit higher than they should be (~700bp). We decided to use the samples that were around ~500bp instead, even though we were expecting a bit higher than that. We made overnights of these samples and mini-prepped them and will be sending them in for sequencing to find out if we have successfully transformed the ligated product.
Week 20: Oct 6 - 10
We sent in the 43 samples for sequencing that we thought contained our BioBricks. The results indicated that the promoter, Pveg, was present in the plasmid, but the rest of the construct was not there. Unlike last time, these samples did not contain CjBlue. This week we made it through two cycles of digestion, ligation, and transformation of our parts. The first round of digestion/ligation/transformation lead to 36 samples likely containing our parts. Samples were then sent in for sequencing. The sequencing results again indicated that the correct promoter (Pveg) was present, but the other pieces of our construct beyond that were not present. We made overnights of all remaining (untested) colonies from the successful plates, but the cPCR failed so we did not go on to prepare these. The second batch of digestion/ligation/transformation we did this week failed as there was no growth at all on the plates. We were using the original synthesized products directly in this restriction digest, instead of putting a small amount it into E. coli and letting the E.coli mass produce it. Therefore, at the end of the week all we had to submit to the iGEM registry was 17 constructs that we had in a different vector. They were unable to be properly inserted into the accepted iGEM vector pSB1C3. We still want people to be able to use these parts that we worked really hard designing for our project so we still posted the information about them on our parts page.
Table 1:Indicates the progress made thus far in BioBricking our parts for the iGEM registry.

Week 21: Oct 13 - 17
Aside from working on content for our Wiki, we attempted to demonstrate the interaction between our reporter and repressor genes. We essentially carried out a double transformation of E. coli. Competent cells were made from E. coli colonies that were initially transformed with the reporter gene, producing red colonies. These competent cells were then transformed with the repressor gene. The reporter gene was carried by a chloramphenicol-resistant plasmid vector and the repressor gene was carried by an ampicillin-resistant plasmid vector, allowing for a reliable double-resistance selection.
The results of our double-transformation showed functional evidence for the interaction between our reporter and repressor genes; we observed a repression of the expression of the RFP reporter. Plates with the two antibiotics produced white colonies indicating that both plasmids were successfully transformed and that RFP was not being expressed. Few red colonies were observed at the edges of the lawn. The occurance of red colonies at the edges are most likely satellite colonies, that is colonies without ampicilin resistance (i.e. without the repressor) which survived due to the breakdown of antibiotic by cells with true ampicilin resistance. Our controls consisted of plating the cells that contain only the reporter gene, and hence only have chloramphenicol resistance, on both chloramphenicol plates and plates with both antibiotics. As expected (Fig. 3)
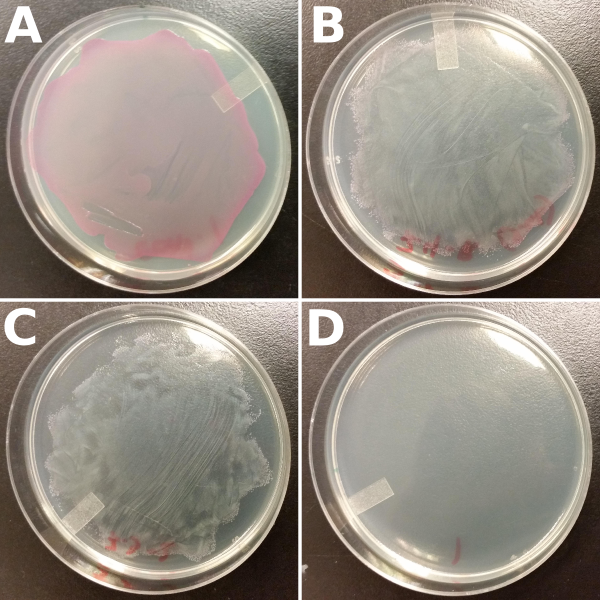
Figure 3: Results of the double-transformation using our reporter and repressor genes. Cells that were double-transformed with both reporter and repressor genes produced white colonies when plated on plates containing both chloramphenicol and ampicilin antibiotics (B and C). Competent cells transformed only once with the reporter gene served as the positive control when plated on chloramphenicol plates (A), and as a negative control when plated on chloramphenicol and ampicilin plates (D).