 "
"
Team:Heidelberg/Software/igemathome
From 2014.igem.org
– iGEM´s software for distributed computing
ABSTRACT
iGEM@home is a platform build upon the Berkeley Open Infrastructure for Network Computing (BOINC). It is supposed to deal with the common problem of insufficient computing resources for iGEM teams. Volunteers download client software that runs computations when their computer is idle. We used the platform to distribute different parts of our Linker Generation program for protein circularization. The most important contribution of our software is the extension of the BOINC framework to support Java and Python applications and the exploration of using distributed computing in the iGEM community.If you want to learn more about the communication platform iGEM@home, please visit the correspondant site in the Human practice section.
Contents |
Introduction
Overview
We, the iGEM Team Heidelberg, were confronted with a similar problem. Working on protein modifications, we wanted to know what effect our modifications could have on the protein by simulating them, before stepping to expensive and time intensive wet lab screenings. Yet iGEM Teams often lack the ability to do resource intensive computations, because they have no access to server clusters. We therefore decided to start iGEM@home, as a universal platform for iGEM-Teams to distribute computing jobs to users all over the world.
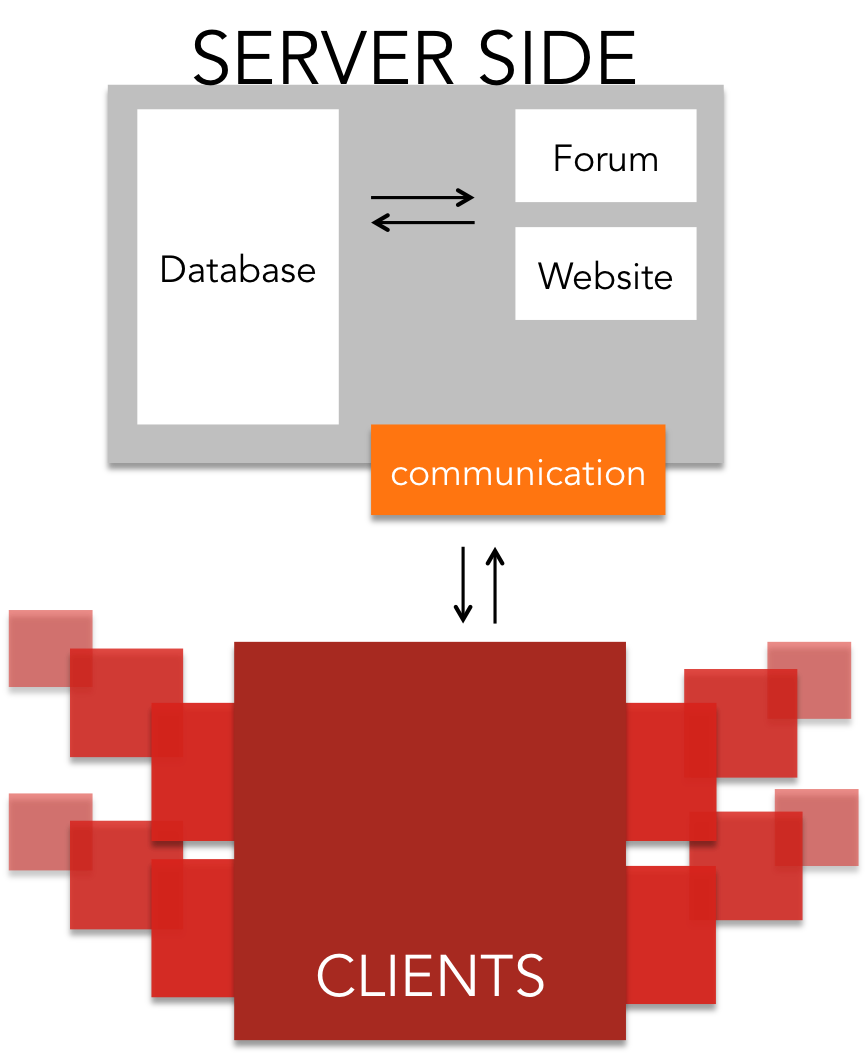
Right now we are using the platform to calculate linkers to enable the simple and efficient circularization of proteins. Further information on the type of computing tasks we are distributing may be found on the Modelling page. In future we hope to see the project grow and prosper when new iGEM teams take over and run their own computations on iGEM@home.
Project Components
The project is separated into two main components:
- Communication related side: The software for the user management, communication between server and client and distribution of jobs
- Scientific calculations: The computing intensive software that is distributed to the volunteers, which run the software and send back the results after completion
Each of these components again is separated into different subcategories.
For the server architecture and the communication between server and client software, we relied on the Berkley Open Infrastructure for Network Computing ([http://boinc.berkeley.edu/trac/wiki/BoincIntro BOINC]). It was created during the development of SETI@home, and has since then been developed further. It includes:
- User management system
- Job management system
- Database
- Backend Interface and forum software
These components were adapted to our needs, for example we added scripts for the easy deployment of our jobs. In addition we implemented a workflow for distributing jobs at multiple levels of the linker generation, which includes waiting for successful results and joining them for the next step.
Scientific calculations:
- The Linker Generator generates a list of all possible linkers that are able to connect the N- and C-terminus of the protein. It sorts out Linkers that cross the protein and then ranks them based on some features of the Linker. In the end it returns a list of the 300 best Linkers for further analysis.
- The server receives the list of possible Linkers and redistributes their pdb’s to the client. Now the second part of the software (Modeller) analyzes the super-secondary structure of the protein and updates the location of the amino acids of the Linker to the place that seems most natural. The pdb with the updated structure is returned to the server.
- The server collects all results and redistributes them to the Linker Evaluator. It analyzes the list of all proteins, calculates an alignment score based on the FatCat Algorithm with the natural protein structure. Furthermore to evaluate the assumptions of the Linker Generator it calculates the length of the linker parts and the angles in between and writes them to a text file that allows the dynamic enhancement of the linker prediction.
The result is a simple file that contains all important information on the process of the Linker Generation and can easily be evaluated on a single laptop.
Implementation Details of the scientific software component:
Python Applications:
As multiple components of our workflow are programmed in python, we needed to find a way to make these python applications portable and executable on computers without python installed, so they can be distributed via the BOINC system. Although python is widely installed on Linux distributions, it is underrepresented on Microsoft Windows operating systems. For the propose of packaging the python script with the required components, we implemented an extraction and version management system in the loader application, which extracts a delivered zip file into a separate folder for each application version and removes old versions if they are no longer needed. Additionally it implements methods for accessing the [http://boinc.berkeley.edu/trac/wiki/BasicApi BOINC-API] via a python accessible “loader” module and file signing checking for input files to avoid licensing issues when using software for academic use only.
Linker Generator:
The linker generator is written in Python. Being one of the most spread programming languages in the scientific community, Python allows to access many modules for the implementation of complex data analysis required for scientific applications. The linker generator relies heavily on the [http://www.numpy.org/ NumPy] module of the Python [http://www.scipy.org/ SciPy] package, which extends the functionality of python to cope with large multi-dimensional arrays and matrices at high computational speed. This is implemented by using compiled C-code and processor specific optimizations for time critical operations. For distribution of the linker generator, we needed to use an alternative approach compared to the distribution of the Modeller software, as there seemed to be incompatibilities between NumPy and the software used for bundling, Nuitka . For further details on the implementation of the Linker Generator or on the principles used for bundling python applications please visit the Linker-Generator or Bundeling Implementation pages.
Modeller:
After talking to multiples experts in the field protein folding analysis, we decided to rely on the state of the art software widely used in this field of research: [Labs Modeller] allows modeling of super secondary structures via homology analysis and discrete energy minimization. Further information on Modeller itself may be found on the Linker Modeling page. To enable the distribution of Modeller we used a software called Nuitka, which defines itself as a python compiler. This means, that the python code is translated into C++-Code which results in a 2-3 fold acceleration of execution speed. Nuitka also allows embedding the complete python runtime into a single executable, thus making the application executable without a python runtime installed. Yet it was required to rewrite parts of the Nuitka software, so the process works flawlessly with the python loader. For further details please visit the implementation page.
Linker Evaluator:
The Linker Evaluator is written in pure Java and usesthe bioinformatics framework BioJava. This is in a bold contrast to the support of the BOINC framework for only the languages C and C++. One of the biggest advantages of Java is that a program can be written and debugged on a single platform and then be run on all the others, which is especially important if one targets a versatile community as with BOINC. To be able to use the Java with BOINC we decided to port the JRE to BOINC. Another approach that was previously discussed in the BOINC community favored the use of Java that is already installed on the computer of the user, but as the number preinstalled JRE’s is declining in the last year we decide to make our program independent of this trend and deploy it as a standalone program.
We therefore utilized the tool javafxpackager that helps bundeling a JRE with a finished java program (JAR file). To incorporate it into the BOINC infrastructure we had to rewrite the Loader program, that is written in C and starts the JVM and executes the actual program.
To reduce the size of the bundle we removed some packages from the JRE, for example all programs that are responsible for displaying graphics (AWT and Swing) and some more files as it was suggested in the JRE Readme (http://www.oracle.com/us/technologies/java/jre-7-readme-430162.html).
The BOINC architecture brings a bunch of methods that are necessary for running applications. Some of these are essential like starting the BOINC process that communicates with the BOINC manager or resolving a file name to the encoded representation that BOINC is internally using. To give users access to these method that were implemented in the C and C++ we had to write a BOINC API Wrapper. It uses the Java Native Interface that gives Java code the ability to call native C and C++ methods. Our BOINCWrapperAPI Java class is distributed with the JRE to give application developers the most natural access to its method and it implements access to the most important BOINC methods. In addition it is easy to extend due to its modularity.
Conclusions
With all this components in place it is very easy to develop applications for BOINC. One can use the popular scripting language Python together with one of the most used science frameworks: SciPy. In addition one can use Java a simple, though powerful fully featured object oriented language and basically all of its libraries to develop applications that can be scaled up from the single development machine to the distribution to thousands of home computers.
With our effort to expand the list of supported languages by the BOINC system with more high level languages, we significantly reduced the implementation effort and required expertise for future BOINC projects and especially for future iGEM teams that want to use iGEM@home and benefit from the available computing power.
References
[1] http://freegamedev.net/wiki/Portable_binaries#System_libraries_that_cannot_be_bundled
[2] http://tldp.org/HOWTO/Program-Library-HOWTO/shared-libraries.html