 "
"
Team:HokkaidoU Japan/Projects/Length
From 2014.igem.org
| (44 intermediate revisions not shown) | |||
| Line 61: | Line 61: | ||
<ul> | <ul> | ||
<li class="ldd_heading"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/Length">Length Variation</a></li> | <li class="ldd_heading"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/Length">Length Variation</a></li> | ||
| - | <li class="ldd_contents"><a href="">Overview</a></li> | + | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/Length">Overview</a></li> |
<li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/Length#Method">Method</a></li> | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/Length#Method">Method</a></li> | ||
<li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/Length#Results">Results</a></li> | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/Length#Results">Results</a></li> | ||
| Line 83: | Line 83: | ||
</ul> | </ul> | ||
<ul> | <ul> | ||
| - | <li class="ldd_heading"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Survey">High | + | <li class="ldd_heading"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Survey">High School</a></li> |
<li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Survey#Education">Education</a></li> | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Survey#Education">Education</a></li> | ||
<li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Survey#Survey">Survey</a></li> | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Survey#Survey">Survey</a></li> | ||
| Line 90: | Line 90: | ||
<li class="ldd_heading"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Discussion">Discussion</a></li> | <li class="ldd_heading"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Discussion">Discussion</a></li> | ||
<li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Discussion#Background">Background</a></li> | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Discussion#Background">Background</a></li> | ||
| - | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Discussion# | + | <li class="ldd_contents"><a href="https://2014.igem.org/Team:HokkaidoU_Japan/Outreach/Discussion#Evaluation">Evaluation</a></li> |
</ul> | </ul> | ||
</div> | </div> | ||
| Line 136: | Line 136: | ||
<div class="fig fig800"> | <div class="fig fig800"> | ||
<img src="https://static.igem.org/mediawiki/2014/8/81/HokkaidoU_length_Gifgifgifff.gif"> | <img src="https://static.igem.org/mediawiki/2014/8/81/HokkaidoU_length_Gifgifgifff.gif"> | ||
| - | <div>Fig. 2 Determining a repression region is | + | <div>Fig. 2 Determining a repression region is difficult.</div> |
</div> | </div> | ||
<p> | <p> | ||
| - | In this project, we made many kinds of anti-sense which repress mRFP, but these anti-sense constructs themselves | + | In this project, we made many kinds of anti-sense which repress mRFP, but these anti-sense constructs themselves are not useful for you. However, in our method, you can create the anti-sense that you desire. We expect this project will help you decide anti-sense sequences. We’d like to tell scientists and iGEMers how easy and accurate to repress the gene expression by anti-sense RNA.</p> |
| - | <h1 style="font-size:43px;" id="Method"> | + | <h1 style="font-size:43px;" id="Method">Methods</h1> |
<p> | <p> | ||
| - | + | We made anti-sense constructs that repress mRFP. The target construct are composed of P<sub>tet</sub> (<a href="http://parts.igem.org/Part:BBa_R0040">BBa_R0040</a>), B0034 (<a href="http://parts.igem.org/Part:BBa_B0034">BBa_B0034</a>), mRFP (<a href="http://parts.igem.org/Part:BBa_E1010">BBa_E1010</a>) and double terminator (<a href="http://parts.igem.org/Part:BBa_B0015">BBa_B0015</a>). | |
| - | As forward primer, | + | |
| + | |||
| + | Insert fragments were synthesized based on BioBrick by PCR. | ||
| + | As forward primer, ”XhoI-P<sub>tet</sub> (-10)”was used for making all fragments. The primer binds to -10 region of P<sub>tet</sub>, and its end has XhoI restriction enzyme site. To change the downstream seqeunce, each reverse primer is designed differently (as90 NcoI, as120 NcoI) (Fig. 3). These primers bind to each specific part of mRFP, and their ends have NcoI restriction enzyme site. By that way, we can get various length of insert fragments, as90 and as120. As90 is the anti-sense that covers 90 bp of mRNA, and as120 is the anti-sense that covers 120 bp of mRNA (complement RBS and a part of mRFP sequence.) Of course, the edges of insert fragments have restriction enzymes XhoI, NcoI sites. | ||
</p> | </p> | ||
| Line 152: | Line 155: | ||
<div class="fig fig800"> | <div class="fig fig800"> | ||
| - | <img src="https://static.igem.org/mediawiki/2014/ | + | <img src="https://static.igem.org/mediawiki/2014/e/ee/HokkaidoU_length_Figfig3.png"> |
| - | <div>Fig. 3 Synthesizing | + | <div>Fig. 3 Synthesizing insert fragments by PCR</div> |
</div> | </div> | ||
| Line 163: | Line 166: | ||
<p> | <p> | ||
| - | After we finished synthesizing insert fragments, we inserted them into our H-stem vector (anti-sense expression vector) by XhoI and NcoI. | + | After we finished synthesizing insert fragments, we inserted them into our H-stem vector (anti-sense expression vector <a href="http://parts.igem.org/Part:BBa_K1524100">BBa_K1524100</a>) by XhoI and NcoI. |
| - | Then, we measured their repression efficiencies. In the same way, we made as30, as60 on H-stem vector | + | Then, we measured their repression efficiencies. In the same way, we made as30, as60 on H-stem vector in other experiment (as30 (<a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/asB0034">Anti-sense B0034</a>) and as60 (<a href="https://2014.igem.org/Team:HokkaidoU_Japan/Projects/H_Stem">H-stem system</a>)). We performed repression experiment by using their four anti-sense constructs. |
</p> | </p> | ||
<div class="clearfix"> | <div class="clearfix"> | ||
</div> | </div> | ||
| - | < | + | <h2>How to assay</h2> |
<p> | <p> | ||
We performed RT-PCR to confirm the transcription of anti-sense RNA (asRNA) constructs.</p> | We performed RT-PCR to confirm the transcription of anti-sense RNA (asRNA) constructs.</p> | ||
<ol> | <ol> | ||
| - | <li> | + | <li>Cultivated the colony in 4 mL LB medium for 16 hours.</li> |
| - | <li> | + | <li>Centrifuged the 4 mL of culture at 10,000 rpm / for 2 min / at 25°C</li> |
| - | <li> | + | <li>Removed the supernatant and add M9ZB medium then voltex the pelet.</li> |
| - | <li> | + | <li>Performed RT-PCR</li> |
| - | <li> | + | <li>Measured absorbance of 260 nm about cDNA.</li> |
</ol> | </ol> | ||
<h1 id="Results">Results</h1> | <h1 id="Results">Results</h1> | ||
<p> | <p> | ||
| - | Though we measured absorbance of | + | Though we measured absorbance of 260 nm about cDNA, we could not get any cDNA. After RNA extraction, we confirmed absorbance of 260 nm (this is the absorbance of nucleic acid). However, after RT-PCR of that products, we could not confirm the existence of nucleic acid. |
<p> | <p> | ||
| - | + | It is presumable that there are some problems in RNA extraction. First, we might have lost RNA during the experiment operation. RNA is degraded easily than DNA because generally RNase can be easily contaminated.</p> | |
<p> | <p> | ||
| - | + | Secondly, the incomplete deactivation of DNase can be considered to be the reason of our failure. If DNase was not deactivated sufficiently, it will result in the degradation of cDNA produced in RT-PCR.</p> | |
<p> | <p> | ||
| - | Though we could not | + | Though we could not make it by wiki freeze, we are going to retry this experiment.</p> |
| Line 201: | Line 204: | ||
<p> | <p> | ||
| - | We theoretically estimated the repression efficiency of asRNA is related to the length | + | We theoretically estimated that the repression efficiency of asRNA is related to its length. To find the optimum length, we must design many lengths of asRNA, and measure their efficiency. However, that takes a lot of time and labor. So, as a future work, we propose an efficient method to synthesize various length of anti-sense.</p> |
| - | <h2> | + | <h2>Methods</h2> |
| - | + | ||
| - | + | ||
| - | + | ||
<h3>Preparation for randomizing</h3> | <h3>Preparation for randomizing</h3> | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| Line 226: | Line 221: | ||
First, we performed PCR on mRFP construct. We call this step "prePCR". We used below primers.</p> | First, we performed PCR on mRFP construct. We call this step "prePCR". We used below primers.</p> | ||
| - | <ul style=" | + | <ul style="padding-left:50px; margin:20px 0;"> |
<li>XhoI-P<sub>tet</sub> (-10)</li> | <li>XhoI-P<sub>tet</sub> (-10)</li> | ||
| - | <li>mRFP | + | <li>mRFP 400 down</li> |
</ul> | </ul> | ||
| - | XhoI-P<sub>tet</sub> (-10) is a primer that binds to -10 region of P<sub>tet</sub> (BBa_R0040), and its | + | XhoI-P<sub>tet</sub> (-10) is a primer that binds to -10 region of P<sub>tet</sub> (BBa_R0040), and its 5’contains XhoI recognition site that is imperative to ligate with our anti-sense vector (H-stem vector). Because it doesn’t contain -35 region, DNA synthesizing starts from P<sub>tet</sub>’s -10 region and PCR products don’t contain a functional part as promoter. |
<br> | <br> | ||
| - | mRFP | + | mRFP 400 down is a primer that binds to mRFP (BBa_E1010) 400 bp downstream. |
<br> | <br> | ||
PCR products that are amplified by these 2 primes showed in Fig. 5. | PCR products that are amplified by these 2 primes showed in Fig. 5. | ||
| Line 246: | Line 241: | ||
<h3>Randomizing</h3> | <h3>Randomizing</h3> | ||
<p> | <p> | ||
| - | + | For this method, we designed two primers shown below. The second primer is a "random primer" which can bind with any sequence on the template DNA. By using these primers, we can synthesize various lengths of anti-sense. | |
| - | + | <ul style="padding-left:50px; margin:20px 0;"> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | <ul style=" | + | |
<li>XhoI-P<sub>tet</sub> (-10)</li> | <li>XhoI-P<sub>tet</sub> (-10)</li> | ||
<li>NcoI-NNNNNN</li> | <li>NcoI-NNNNNN</li> | ||
</ul> | </ul> | ||
| + | </p> | ||
<p> | <p> | ||
| - | + | However, the random primer only has 6 nt of region that anneals with the template DNA. Eventually, the Tm value of this primer gets extremely low. This causes some difficulties to PCR. As a solution, we have to take three unusual steps for PCR. | |
<br> | <br> | ||
| - | + | First, to single-strandize the template DNA, putting it at 95°C / for 3 min and then putting it in 0°C water. By this procedure, the template DNA will be kept denatured in low temperature. | |
| + | <br>Second, to anneal the primer and amplify the sequences in low temperature, we use "Klenow Fragment", a polymerase whose optimum temperature is 37°C. In this step, we can not amplify fragments, but can make templates of various lengths. | ||
| + | <br>In the third step, we amplify the templates we mentioned above at 68°C just like the usual PCR. </p> | ||
<div class="fig fig400" > | <div class="fig fig400" > | ||
<img src="https://static.igem.org/mediawiki/2014/f/f5/HokkaidoU_length_Random3.png"> | <img src="https://static.igem.org/mediawiki/2014/f/f5/HokkaidoU_length_Random3.png"> | ||
| - | <div>Fig. | + | <div>Fig. 6 How to make random anti-sense</div> |
</div> | </div> | ||
| - | |||
<p> | <p> | ||
| - | + | By these methods, you can get various lengths of asRNA of target gene. These fragments are all in one mixture of PCR products. Thus, by digesting, ligating the fragments into pHN1257 vector (published by Nakashima) and transforming them to cells, you can get various colonies with different lengths of anti-sense. The colony with the optimum length of anti-sense must be on the plate!</p> | |
| - | </p | + | |
| - | + | ||
| - | + | ||
| - | < | + | <h2>Results</h2> |
<p> | <p> | ||
| - | We | + | We carried out this plan into practice. We used mRFP as the target gene. After prePCR, we performed PCR by following the steps we described above. The protocol of PCR is shown below. |
| - | + | ||
| - | We used | + | |
<div class="fig fig800"> | <div class="fig fig800"> | ||
| - | <img src="https://static.igem.org/mediawiki/2014/ | + | <img src="https://static.igem.org/mediawiki/2014/2/2c/HokkaidoU_length_Last_recipe.png"> |
| - | <div>Fig. | + | <div>Fig. 7 Randomizing protocol</div> |
| + | </div> | ||
| + | |||
| + | <div class="clearfix"> | ||
</div> | </div> | ||
| - | |||
<div class="fig fig400" > | <div class="fig fig400" > | ||
| - | <img src="https://static.igem.org/mediawiki/2014/ | + | <img src="https://static.igem.org/mediawiki/2014/4/4c/HokkaidoU_length_Protocols.png"> |
| - | <div>Fig. | + | <div>Fig. 8 Results of randomizing</div> |
</div> | </div> | ||
<p> | <p> | ||
<! | <! | ||
<p> | <p> | ||
| - | + | We performed an experiment in two protocols, A and B. The annealing temperature and PCR cycles are different beteween A and B. We inserted them into vectors and transformed them to cells. We got some colonies in both plates! More colonies appeared in Protocol B than Protocol A. After that, we performed colony PCR. However, we could not get the result of colony PCR. | |
| - | + | ||
</p> | </p> | ||
<div class="clearfix"> | <div class="clearfix"> | ||
| Line 311: | Line 292: | ||
<h2>Discussion</h2> | <h2>Discussion</h2> | ||
<p> | <p> | ||
| - | + | Getting this result that the colony PCR had failed, we stopped this project. However, recently, we realized that the stem sequences have difficuties in PCR (according to Mr. Nakashima). Therefore, we cannot conclude that this project have completely failed.</p> | |
| - | + | ||
| - | < | + | <h3>Reference</h3> |
| - | < | + | |
<ol class="citation-list"> | <ol class="citation-list"> | ||
| - | <li id="cite-1">N Nakashima <i>et al.</i> (2006) Paired termini stabilize antisense RNAs and enhance conditional gene silencing in <i>Escherichia coli</i>. Nucleic Acids Res 34: 20 e138</li> | + | <li id="cite-1">N. Nakashima <i>et al.</i> (2006) Paired termini stabilize antisense RNAs and enhance conditional gene silencing in <i>Escherichia coli</i>. Nucleic Acids Res 34: 20 e138</li> |
</ol> | </ol> | ||
</div> | </div> | ||
| Line 330: | Line 309: | ||
<div id="footer-content"> | <div id="footer-content"> | ||
<div id="footer-logo"> | <div id="footer-logo"> | ||
| - | <a href="http://igemhokkaidou.com | + | <a href="http://igemhokkaidou.wordpress.com"><img style="height:150px;position:relative;" src="https://static.igem.org/mediawiki/2014/3/39/HokkaidoU_logo_transparent.png"></a> |
</div> | </div> | ||
<div id="footer-twitter"> | <div id="footer-twitter"> | ||
Latest revision as of 15:21, 9 September 2015


Overview
Using anti-sense RNA (asRNA) is one of the methods to repress gene expression. It is known that the length of anti-sense sequence is related to its repression efficiency (N. Nakashima et al., 2006[1]), but the details of the relation are still unclear. In this project, we made different lengths of anti-sense sequence (Fig. 1).

Our experiments will be a clue for other iGEMers who want to design their own anti-sense sequence.
Introduction
In repressing gene by anti-sense RNA, it is important to determine the length of anti-sense. However, it is difficult to do it. Theoretically, if the length is too long, it doesn’t repress target RNA effectively. The reason is because the RNA polymerase takes a lot of time to synthesize them, and the diffusion rate of them also gets low. However, too short asRNA also has some problems. The short anti-sense cannot bind to the specific part of mRNA because it has too short complementary sequences of target RNA. In the industrial and academic fields, people hope to use anti-sense that has suitable repression efficiency. For example, you can create knock down recombinant organisms easily by using strong anti-sense, and in iGEM, you can make bio-devices which have a complicated gene network and require fine-tuned gene expression. Gene expression is not only ON or OFF. As stated above, each case needs each repression efficiency. Researchers currently tried to change anti-sense repression efficiency by changing anti-sense’s binding sequence. However, it is known that this method is difficult.

In this project, we made many kinds of anti-sense which repress mRFP, but these anti-sense constructs themselves are not useful for you. However, in our method, you can create the anti-sense that you desire. We expect this project will help you decide anti-sense sequences. We’d like to tell scientists and iGEMers how easy and accurate to repress the gene expression by anti-sense RNA.
Methods
We made anti-sense constructs that repress mRFP. The target construct are composed of Ptet (BBa_R0040), B0034 (BBa_B0034), mRFP (BBa_E1010) and double terminator (BBa_B0015). Insert fragments were synthesized based on BioBrick by PCR. As forward primer, ”XhoI-Ptet (-10)”was used for making all fragments. The primer binds to -10 region of Ptet, and its end has XhoI restriction enzyme site. To change the downstream seqeunce, each reverse primer is designed differently (as90 NcoI, as120 NcoI) (Fig. 3). These primers bind to each specific part of mRFP, and their ends have NcoI restriction enzyme site. By that way, we can get various length of insert fragments, as90 and as120. As90 is the anti-sense that covers 90 bp of mRNA, and as120 is the anti-sense that covers 120 bp of mRNA (complement RBS and a part of mRFP sequence.) Of course, the edges of insert fragments have restriction enzymes XhoI, NcoI sites.


After we finished synthesizing insert fragments, we inserted them into our H-stem vector (anti-sense expression vector BBa_K1524100) by XhoI and NcoI. Then, we measured their repression efficiencies. In the same way, we made as30, as60 on H-stem vector in other experiment (as30 (Anti-sense B0034) and as60 (H-stem system)). We performed repression experiment by using their four anti-sense constructs.
How to assay
We performed RT-PCR to confirm the transcription of anti-sense RNA (asRNA) constructs.
- Cultivated the colony in 4 mL LB medium for 16 hours.
- Centrifuged the 4 mL of culture at 10,000 rpm / for 2 min / at 25°C
- Removed the supernatant and add M9ZB medium then voltex the pelet.
- Performed RT-PCR
- Measured absorbance of 260 nm about cDNA.
Results
Though we measured absorbance of 260 nm about cDNA, we could not get any cDNA. After RNA extraction, we confirmed absorbance of 260 nm (this is the absorbance of nucleic acid). However, after RT-PCR of that products, we could not confirm the existence of nucleic acid.
It is presumable that there are some problems in RNA extraction. First, we might have lost RNA during the experiment operation. RNA is degraded easily than DNA because generally RNase can be easily contaminated.
Secondly, the incomplete deactivation of DNase can be considered to be the reason of our failure. If DNase was not deactivated sufficiently, it will result in the degradation of cDNA produced in RT-PCR.
Though we could not make it by wiki freeze, we are going to retry this experiment.
Developmental experiment
We theoretically estimated that the repression efficiency of asRNA is related to its length. To find the optimum length, we must design many lengths of asRNA, and measure their efficiency. However, that takes a lot of time and labor. So, as a future work, we propose an efficient method to synthesize various length of anti-sense.
Methods
Preparation for randomizing

Before randomizing, we have to perform some steps to make the effective anti-sense fragments. First, we performed PCR on mRFP construct. We call this step "prePCR". We used below primers.
- XhoI-Ptet (-10)
- mRFP 400 down
mRFP 400 down is a primer that binds to mRFP (BBa_E1010) 400 bp downstream.
PCR products that are amplified by these 2 primes showed in Fig. 5.
Through this step, we can get insert fragments containing SD (Shine-Dargalno) sequence and start codon that are important to effictive repression.
Randomizing
For this method, we designed two primers shown below. The second primer is a "random primer" which can bind with any sequence on the template DNA. By using these primers, we can synthesize various lengths of anti-sense.
- XhoI-Ptet (-10)
- NcoI-NNNNNN
However, the random primer only has 6 nt of region that anneals with the template DNA. Eventually, the Tm value of this primer gets extremely low. This causes some difficulties to PCR. As a solution, we have to take three unusual steps for PCR.
First, to single-strandize the template DNA, putting it at 95°C / for 3 min and then putting it in 0°C water. By this procedure, the template DNA will be kept denatured in low temperature.
Second, to anneal the primer and amplify the sequences in low temperature, we use "Klenow Fragment", a polymerase whose optimum temperature is 37°C. In this step, we can not amplify fragments, but can make templates of various lengths.
In the third step, we amplify the templates we mentioned above at 68°C just like the usual PCR.

By these methods, you can get various lengths of asRNA of target gene. These fragments are all in one mixture of PCR products. Thus, by digesting, ligating the fragments into pHN1257 vector (published by Nakashima) and transforming them to cells, you can get various colonies with different lengths of anti-sense. The colony with the optimum length of anti-sense must be on the plate!
Results
We carried out this plan into practice. We used mRFP as the target gene. After prePCR, we performed PCR by following the steps we described above. The protocol of PCR is shown below.
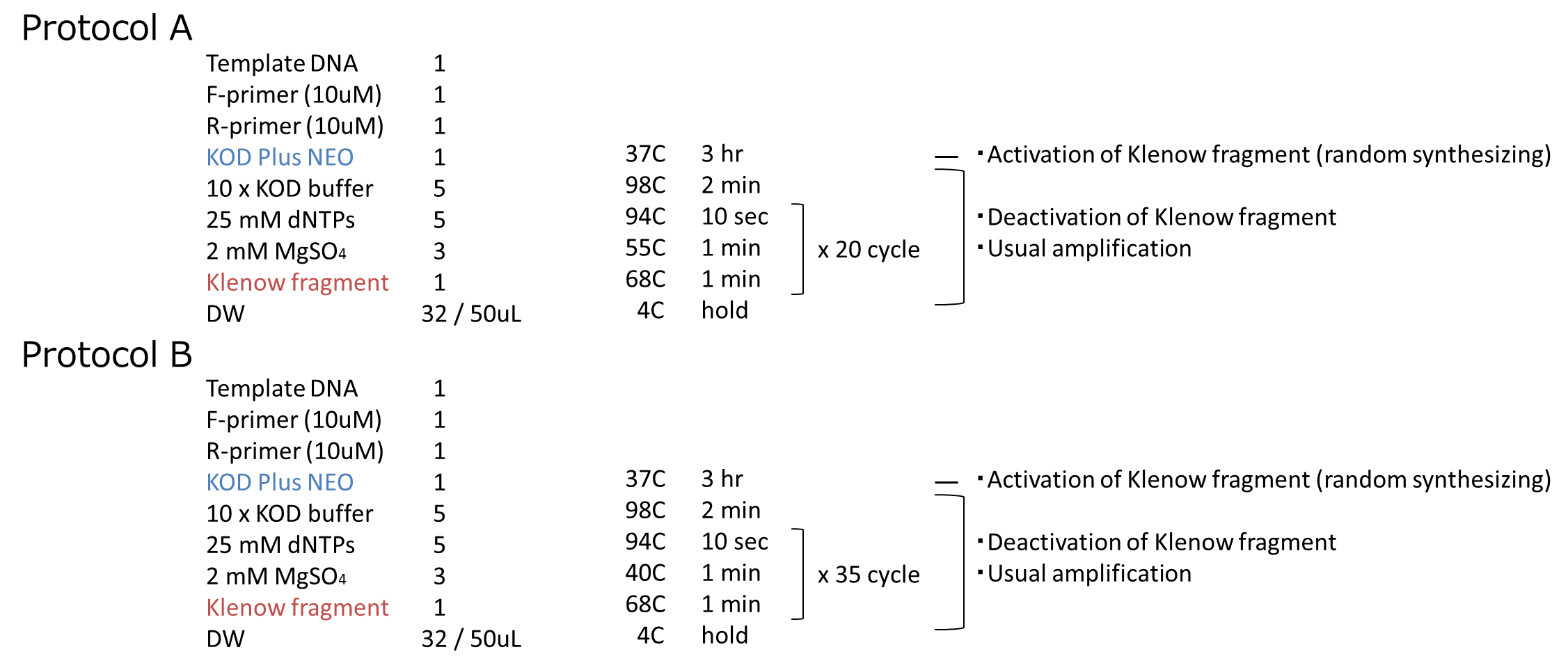

We performed an experiment in two protocols, A and B. The annealing temperature and PCR cycles are different beteween A and B. We inserted them into vectors and transformed them to cells. We got some colonies in both plates! More colonies appeared in Protocol B than Protocol A. After that, we performed colony PCR. However, we could not get the result of colony PCR.
Discussion
Getting this result that the colony PCR had failed, we stopped this project. However, recently, we realized that the stem sequences have difficuties in PCR (according to Mr. Nakashima). Therefore, we cannot conclude that this project have completely failed.
Reference
- N. Nakashima et al. (2006) Paired termini stabilize antisense RNAs and enhance conditional gene silencing in Escherichia coli. Nucleic Acids Res 34: 20 e138
