 "
"
Team:IvyTech SouthBend IN/progress
From 2014.igem.org
(Difference between revisions)
| (3 intermediate revisions not shown) | |||
| Line 69: | Line 69: | ||
<li><a href="#929104">9/29/14-10/4/14</a></li> | <li><a href="#929104">9/29/14-10/4/14</a></li> | ||
<li><a href="#10511">10/5/14-10/11/14</a></li> | <li><a href="#10511">10/5/14-10/11/14</a></li> | ||
| + | <li><a href="#101218">10/12/14-10/18/14</a></li> | ||
<li><a href="#DR">Data Review 10/14/14</li> | <li><a href="#DR">Data Review 10/14/14</li> | ||
</ul> | </ul> | ||
| Line 96: | Line 97: | ||
</div> | </div> | ||
| - | <div class="ivyleft"> | + | <div class="ivymask rightmenu"> |
| + | |||
| + | <div class="ivyleft"> | ||
<div class="ivy1"> | <div class="ivy1"> | ||
| + | <!-- column 1 start --> | ||
| + | <div id="memo3g"><a name="101218">Week</a> of 10/12/14 through 10/18/14</div><br /> | ||
| + | <ul> | ||
| + | <li>Our goal was to create a microfluidic device. We used negative lithography to create a patterned wafer and the pattern in the wafer will be used as a negative master mold to create a PDMS microfluidic structure. The PDMS microfluidic structure will use the application of hydrostatic transport of a liquid media. The PDMS microfluidic structure is being adhered to a glass slide to create a sealed chamber and that will be used to transport liquids through microfluidic channels. A microfluidic channel is commonly defined as having one or more dimension less than 100 microns in size. Transport mechanisms used by microfluidics to transfer liquids include capillary forces, hydrostatic pressure gradients, electrokinetics, pumps, magnetism and digital arrays. Microfluidic channel material is important to consider when choosing the transport mechanism.</li><br /> | ||
| + | <li>The goal of the experiment is to create a master mold with a patterned wafer, to use that master mold to create a PDMS microfluidic device and to transport liquids through the PDMS microfluidic device. </li><br /> | ||
| + | <li>The wafers are now primed for photoresist application. The photoresist being used is SU8-25 which should give a thickness of approximately 25 microns. We decided to try to increase the thickness of the resist to 75-80 microns by slowing the spin rate to 1000 rpm and applying the resist extra thick. Room lights are turned off and special orange lights that filter out light below 530 nanometers are used because SU8-25 photoresist is sensitive to ultraviolet light. The SU8-25 photoresist is applied using a static spin. One at a time each wafer is placed on a two inch chuck and a centering spin is done to ensure the wafer is centered. An eight milliliter pipette is used to apply the SU8-25 photoresist to the wafer. The pipette is used to cover the entire wafer with SU8-25 photoresist and to remove any air bubbles. The first step of the recipe is a 5 second dwell at 500 RPM with a ramp rate of 100 RPM per second. The second step of the recipe is a 40 second dwell at 1000 RPM, we changed the exposure time from 45 to 60 seconds. A soft bake on a hot plate is performed on the wafer (after the photoresist was spun on) to remove any solvents and harden the photoresist. The wafer’s soft bake is started at 65° C and after three minutes the hot plate temperature is raised to 95° C for an additional seven minutes of baking. </li><br /> | ||
| + | <li>We then performed a post exposure bake for 1min at 65 ° C then raised the hot plate to 95 ° C for additional 3 minutes. After PEB the wafer is developed by placing it in a bath of developer and gently swirled for five minutes to develop the pattern in each wafer. The wafers are removed from the bath, rinsed with isopropanol and air dried. The wafers are then brought back down to the nanotech lab and images are taken of them with the optical microscope. We the measured the thickness on the profilometer and found we hit our mark at 80,162 Å .</li><br /> | ||
| + | <li>The procedure we used came from a microfluidics nanotechnology negative lithography lab. In preparing the wafers they are cleaned with acetone, isopropanol, deionized water and dried with an air can. The wafers go through a dehydration bake on a hot plate at 110° C for three to four minutes to remove any residual liquids. A HMDS application is performed on the wafers to improve photoresist adhesion to the wafer substrate. HMDS is applied to the wafers by putting them into a sealed but non-vacuumed chamber with an open bottle of HDMS for 15 minutes. After removal from the chamber the wafers go through another dehydration bake on a hot plate for 15 seconds at 110° C. </li></br> | ||
| + | </ul> | ||
| + | <!-- column 1 end --> | ||
| + | </div> | ||
| + | <div class="ivy2"> | ||
| + | <!-- column 2 start --> | ||
| + | <p><center><img src="http://i.imgur.com/vIsgpcX.png" style="width: 100%;max-height: 100%" /></center></p> | ||
| + | <!-- column 2 end --> | ||
| + | </div> | ||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | <div class="ivymask leftmenu"> | ||
| + | |||
| + | <div class="ivyleft"> | ||
| + | <div class="ivy1"> | ||
| + | <!-- column 1 start --><br /> | ||
| + | <div id="memo2g"><a name="92128">Week</a> of 10/5/14 through 10/11/14</div> | ||
| + | <ul> | ||
| + | <li>This week we began work on the nanotechnology aspects of our project. The main focus was working on the microfluidic channel, the volume of space that is going to hold the water sample while the biochemical reaction for beta-galactosidase is taking place. We used negative lithography to cast a master mold of a chamber we designed and thought would be best suited for our device. After the mold was cast and sent through numerous bake sessions, it was inspected and determined to be an acceptable mold. We then made a polydimethylsiloxane (PDMS) mixture with a 10:1 ratio with a curing agent. After pouring it into the mold, great care was taken to ensure there were no bubbles left in the PDMS mixture. After being left to dry and hardened, a plasma generator was used to bind the cut out piece to a glass slide. The patterned wafer negative mold microfluidic chamber width is 14.562mm. Its height is 21.3μm at the sidewall and 22.3μm in the middle. A single pillar width is 1.611mm and “height” is 20.8μm. The three microfluidic channel widths are 0.6688mm, 0.5472mm and 0.6080mm while the middle microfluidic channel height is 20.9μm. It can hold a water sample of 6.75 uL. Unfortunately, after testing it with ink we got negative results. Two chambers were tested and both collapsed. We are going to try again using shorter channels and combining pillars so there is a better support system. </li><br /> | ||
| + | </ul> | ||
| + | <!-- column 1 end --> | ||
| + | </div> | ||
| + | <div class="ivy2"> | ||
| + | <!-- column 2 start --> | ||
| + | <p><center><img src="http://i.imgur.com/v78FAXF.png" style="width: 75%;max-height: 75%" /></center></p> | ||
| + | <!-- column 2 end --> | ||
| + | </div> | ||
| + | </div> | ||
| + | </div> | ||
| - | <div class="ivymask | + | <div class="ivymask rightmenu"> |
<div class="ivyleft"> | <div class="ivyleft"> | ||
<div class="ivy1"> | <div class="ivy1"> | ||
Latest revision as of 23:12, 17 October 2014



Progress by Week
- 5/25/14-5/31/14
- 6/15/14-6/21/14
- 6/22/14-6/28/14
- 6/29/14-7/5/14
- 7/6/14-7/12/14
- 7/13/14-7/19/14
- 7/20/14-7/26/14
- 7/27/14-8/2/14
- 8/3/14-8/9/14
- 8/10/14-8/16/14
- 8/17/14-8/23/14
- 8/24/14-8/30/14
- 8/31/14-9/6/14
- 9/7/14-9/13/14
- 9/14/14-9/20/14
- 9/21/14-9/28/14
- 9/29/14-10/4/14
- 10/5/14-10/11/14
- 10/12/14-10/18/14
- Data Review 10/14/14
Data Review 10/14/14

- Data from a replication of Stanick’s assay with K1477014: Cells were grown overnight in LB or LB/glucose. Cells in LB were stimulated with 200 uM IPTG for 1 hour and then all cell cultures were divided into 1mL aliquots to which calcium chloride and magnesium sulfate were added. T7 bacteriophage (1 x 10^5 pfu) was added to the appropriate culture and the absorbance at A600 was followed until virus lysis was evident. Cell cultures were then transferred to SpinX filter devices and the media was separated from the cells. Aliquots of filtrates from cells only, T7-treated cultures, or from T7-treated culture were transferred to a microtiter plate. Buffer Z with 2 mercaptoethanol was added and absorbance at 415 nm was determined after 30 minutes.
- Results: Both E.coli C and K1477014 cells stimulated with IPTG “leaked” β-gal activity (Cells only). Top10 cells were negative as expected. β-gal activity was suppressed in all cell lines by glucose as expected. T7 treatment resulted in an increased release of β-gal activity from both E. coli C and K1477014. The combination of T7 lysates from Top10 cells stimulated with IPTG and K1477014 cells grown in glucose did not show any β-gal activity. .
- Conclusion: Cells stimulated with IPTG and subjected to this protocol release β-gal activity. This might be due to β-gal activity that accumulated in the media as the cells grew. Another possibility is that β-gal activity was released when the calcium chloride or magnesium sulfate solutions were added before virus lysis or perhaps released during centrifugation of the cells in the SpinX columns.
- Most important however is that β-gal activity was found in the medium of lysed and unlysed K1477014 suggesting that the LacZ alpha and omega polypeptides stay combined once they are released! That is great news!
- T7 treatment caused an increase in the release of β-gal enzyme from the E. coli C and the β-gal enzyme (alpha-omega polypeptide combo) from K1477014.
- Unfortunately, the combination of T7 lysates of Top 10 cells treated with IPTG, which should have formed LacZ omega polypeptides and the K1477014 cells treated with glucose, which should have formed only LacZ alpha polypeptides, did not have any detectable β-gal activity.
- This lack of activity could be due to the slow association of the alpha and omega fragments, maybe slowed by the Buffer Z and calcium and magnesium salts or, worse, it means that the alpha and omega polypeptides have to associate inside a cell to be joined into a working enzyme. We can study this further by letting the combination of fragments incubate together for hours in hopes that β-gal activity appears. If it does that is great because that is the proof of principle that we are after.
Week of 10/12/14 through 10/18/14
- Our goal was to create a microfluidic device. We used negative lithography to create a patterned wafer and the pattern in the wafer will be used as a negative master mold to create a PDMS microfluidic structure. The PDMS microfluidic structure will use the application of hydrostatic transport of a liquid media. The PDMS microfluidic structure is being adhered to a glass slide to create a sealed chamber and that will be used to transport liquids through microfluidic channels. A microfluidic channel is commonly defined as having one or more dimension less than 100 microns in size. Transport mechanisms used by microfluidics to transfer liquids include capillary forces, hydrostatic pressure gradients, electrokinetics, pumps, magnetism and digital arrays. Microfluidic channel material is important to consider when choosing the transport mechanism.
- The goal of the experiment is to create a master mold with a patterned wafer, to use that master mold to create a PDMS microfluidic device and to transport liquids through the PDMS microfluidic device.
- The wafers are now primed for photoresist application. The photoresist being used is SU8-25 which should give a thickness of approximately 25 microns. We decided to try to increase the thickness of the resist to 75-80 microns by slowing the spin rate to 1000 rpm and applying the resist extra thick. Room lights are turned off and special orange lights that filter out light below 530 nanometers are used because SU8-25 photoresist is sensitive to ultraviolet light. The SU8-25 photoresist is applied using a static spin. One at a time each wafer is placed on a two inch chuck and a centering spin is done to ensure the wafer is centered. An eight milliliter pipette is used to apply the SU8-25 photoresist to the wafer. The pipette is used to cover the entire wafer with SU8-25 photoresist and to remove any air bubbles. The first step of the recipe is a 5 second dwell at 500 RPM with a ramp rate of 100 RPM per second. The second step of the recipe is a 40 second dwell at 1000 RPM, we changed the exposure time from 45 to 60 seconds. A soft bake on a hot plate is performed on the wafer (after the photoresist was spun on) to remove any solvents and harden the photoresist. The wafer’s soft bake is started at 65° C and after three minutes the hot plate temperature is raised to 95° C for an additional seven minutes of baking.
- We then performed a post exposure bake for 1min at 65 ° C then raised the hot plate to 95 ° C for additional 3 minutes. After PEB the wafer is developed by placing it in a bath of developer and gently swirled for five minutes to develop the pattern in each wafer. The wafers are removed from the bath, rinsed with isopropanol and air dried. The wafers are then brought back down to the nanotech lab and images are taken of them with the optical microscope. We the measured the thickness on the profilometer and found we hit our mark at 80,162 Å .
- The procedure we used came from a microfluidics nanotechnology negative lithography lab. In preparing the wafers they are cleaned with acetone, isopropanol, deionized water and dried with an air can. The wafers go through a dehydration bake on a hot plate at 110° C for three to four minutes to remove any residual liquids. A HMDS application is performed on the wafers to improve photoresist adhesion to the wafer substrate. HMDS is applied to the wafers by putting them into a sealed but non-vacuumed chamber with an open bottle of HDMS for 15 minutes. After removal from the chamber the wafers go through another dehydration bake on a hot plate for 15 seconds at 110° C.

Week of 10/5/14 through 10/11/14
- This week we began work on the nanotechnology aspects of our project. The main focus was working on the microfluidic channel, the volume of space that is going to hold the water sample while the biochemical reaction for beta-galactosidase is taking place. We used negative lithography to cast a master mold of a chamber we designed and thought would be best suited for our device. After the mold was cast and sent through numerous bake sessions, it was inspected and determined to be an acceptable mold. We then made a polydimethylsiloxane (PDMS) mixture with a 10:1 ratio with a curing agent. After pouring it into the mold, great care was taken to ensure there were no bubbles left in the PDMS mixture. After being left to dry and hardened, a plasma generator was used to bind the cut out piece to a glass slide. The patterned wafer negative mold microfluidic chamber width is 14.562mm. Its height is 21.3μm at the sidewall and 22.3μm in the middle. A single pillar width is 1.611mm and “height” is 20.8μm. The three microfluidic channel widths are 0.6688mm, 0.5472mm and 0.6080mm while the middle microfluidic channel height is 20.9μm. It can hold a water sample of 6.75 uL. Unfortunately, after testing it with ink we got negative results. Two chambers were tested and both collapsed. We are going to try again using shorter channels and combining pillars so there is a better support system.

Week of 9/29/14 through 10/4/14
- This week our team had to split up. Two of our members-Kevin and Lyuda- went along with our adviser Professor Twaddle to Posters on the Hill for CCURI (Community College Undergraduate Research Initiative) in Washington D.C. While they were away, Karinne worked on subculturing into fresh broth so that we can make sure to have clean cell lines to work with.
- Also, after an exchange of emails with the Carnegie Melon iGEM team we realized we would be able to help test a beta kit of theirs that make the DNA of wheat germ visible after only a few simple steps. It was easy to use and easy to understand and we thank the team profusely for the fun experience of this experiment!
- We also started delving into the nanotech side of our project by running a protocol of various monolayers to test adhesion for the most favorable surface for biofilm adhesion. We are doing this to make a more optimal environment for the lyophilized cells that will be kept in the chamber of our device until a sample is introduced. 2% solution in acetone was used. After monolayers were added to slides they were incubated while being soaked in broth cultures of Top 10 E. coli cells. (3-Aminopropyl)triethoxysilane (APTES) was found to have the most cells attached when counting results under a microscope.
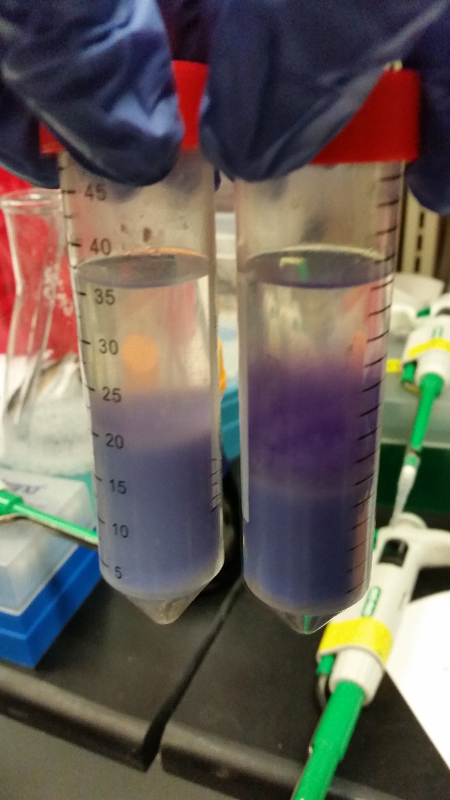
Week of 9/21/14 through 9/28/14
- This week we are going to keep working on our assay of Beta-galactosidase activity. So far we have had two unsuccessful attempts at our run-through of it with intact cells. This week we started it a third and fourth time and had success! Our E. coli C turned to a red color along with our K1477014 part. Not only that, but our negative control stayed negative. Everything worked out perfectly except for the time elapsed to get these results. We expected it to take 12-14 hours for a color change to occur, but it ended up taking about 36 hours in one instance and a little over 48 hours in the last attempt. However we are still pleased to have achieved success (we are still fighting contamination.)
- We also did two runs of this assay with chemically lysed cells. In the first run through, we got positive results. However, an inquiry led to the discovery that one of the chemicals used could potentially denature any enzymes present, and so we had to throw out that data. The second time we used an appropriate chemical, but had positive results on everything-including the negative control. We are now out of our required materials for chemically lysing cells.
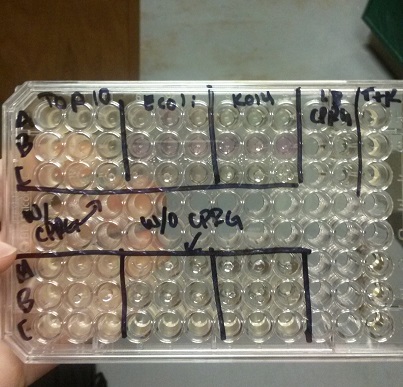
Week of 9/14/14 through 9/20/14
- This week we began our attempts on the Beta-galactosidase activity assay. We plan to run this assay three different ways. We will first complete it using intact cells, measuring the activity that is produce inside the cell. Then we will use chemically lysed cells. By lysing the cells in this way, we are ensuring a higher accuracy in regards to amount of cells being lysed. The chemicals will destroy the cell wall and the beta-gal complementation can occur in vitro.
- Finally, we plan to bring the core idea of our research project together, and then we will run this assay using cells that have been lysed with bacteriophage.
- In all of these, we will be using chlorophenol red (CPRG) as the substrate. This will cause a color change that ranges from yellow to dark red. We are using Top 10 E. coli cells as our negative control. These only produce one out of the two parts that make up the enzyme. We will be using E. coli C as our positive control. We are testing the activity produced in our part K1477014 transformed into Top 10 cells.
- Our first try did not yield correct results, with all three cell samples leading to a positive color change. Our second attempt also did not turn out right. Only the E. coli C gave a positive reading of Beta-gal activity.

Week of 9/7/14 through 9/13/14
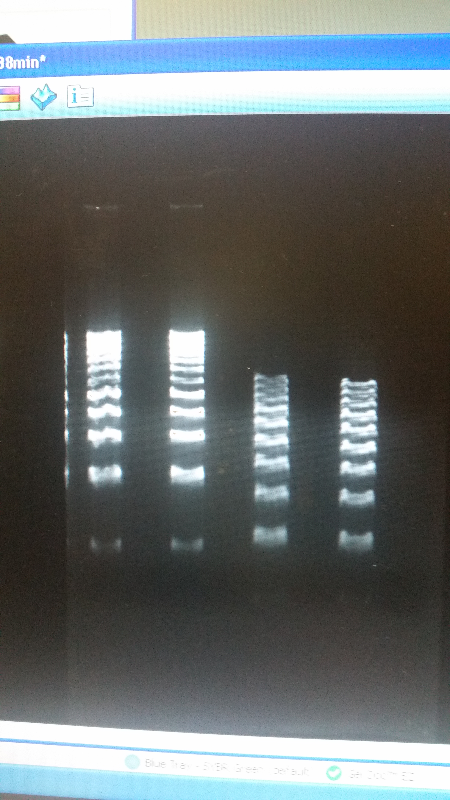
- Continuing in our goal towards collaboration with other iGEM teams, we came into contact with the Cornell iGEM team. They are working on detecting heavy metal pollution in water. To aid them in their research, we sent them two 50cc water samples from local sources (including the Saint Joseph river and Potato Creek.) Hopefully this helped them out!
- As well as our journey towards working with other teams, this week we reverted back to running an electrophoresis gel for our parts. With the help of a new addition to our advisers-Shaunasee Kocen-we found newer materials, and began using different sized DNA ladders to compare. After a few more tries, we were able to get our DNA bands looking great, and we are ready to send our parts into the iGEM registry.
- Left image is our part, K1477014. Right image is our part, K1477030



Week of 8/24/14 through 8/30/14
- This week our team's persistence and effort came to fruition when we finally achieved a virus titer plaque assay that turned out perfectly! There was no contamination. The cell growth did not overpower the working virus bacteriophage. Best of all, we have clear dead zones that show consistency with the corresponding viral dilutions. We are now ready to begin performing the Beta-galactosidase activity assay.


Week of 8/17/14 through 8/23/14
- We tried again to get good results of the virus titer plaque assay. We are still getting a bit of contamination, although the growth is not bright yellow as the rest of the contamination has been. This leads us to believe that we are still using a concentration of cells that is too high. We have been using Top 10 E. coli cells with an A600 reading of ~0.400. We are now going to start using a broth culture of cells with an A600 reading of ~0.200.


Week of 8/10/14 through 8/16/14
- During this week we restocked our supplies for broth and agar growth media, as we were running very low.
- One of our advisers suggested we change the buffer we were using for our gel electrophoresis, and that when preparing the DNA mixture to insert into the gel, we should use buffer instead of DDI water, since this is what is present when the gel is running.
- However, even after changing the buffer, we did not experience any better results. We tried a few times. First, the DNA of our parts did not show up at all. And again, our DNA ladder did not show up correctly.
Week of 8/3/14 through 8/9/14
- During this week, the team realized that the contamination was coming from our stock of cells that was being kept in the -80C freezer. We have no idea how they became contaminated, seeing as continued to use the line of cells we froze for a week after we froze them, but before any contamination occurred. Nonetheless, one of our advisers has procured a new stock of top 10 E. coli cells for us to use.
- After unsuccessful runs of electrophoresis gels, we have run out of DNA to try to measure. So, we took some time out this week to streak, grow, and purify lines of our K1477014 and K1477030 parts on both pSB1C3 and pSB1AT3 backbones.
Week of 7/27/14 through 8/2/14
- After talking with an outside adviser, we have realized that much of our protocol was wrong when it came to running an electrophoresis gel. We were doing a "quick run" protocol, running a high voltage for a short time. Now we are running at a significantly lower voltage, and tripling the time it takes to complete the gel. The DNA bands to our parts are now turning out well. However, the bands in the DNA ladder still look smashed together, which continues to make it impossible to count our parts' kilobases.
- Our team has started working on a plaquing assay with T7 and T4 viruses. We are trying to figure out exactly what concentration of virus we need to work with. Unfortunately, the problem of contamination has become an even bigger issue during the performance of this protocol. The outcome yields a lawn growth of cells that is too thick to make out sufficient plaques of dead zones and there are bright yellow colonies found on a few of the plates (Five plates total in all.) We worry also that perhaps we are using a broth culture of top 10 E.coli cells whose concentration may be too high for the virus to kill.
Week of 7/20/14 through 7/26/14
- We have created a stock of our two parts on the pSB1C3 backbone. We purified both parts, will prepare to send them in to iGEM by running an electrophoresis gel to prove that our parts are what we say they are. We ran two gels, but the DNA bands are coming out deformed for some reason and the DNA ladder does not have distinguished bands that can be used to count how many kilo-base pairs our part has. We are looking into why this is. Possible explanations include incorrect voltage, incorrect agarose gel thickness, or incorrect amount of DNA added. Adjustments will be made in the future.
- We have also performed 3A biobrick assembly two more times and have a new stock of our part on its original backbone pSB1AT3.
- Our problem of contamination is continuing to persist. During our attempts of recreating a new stock of our parts, we found bright yellow colonies all over more than half of the plates we streaked.

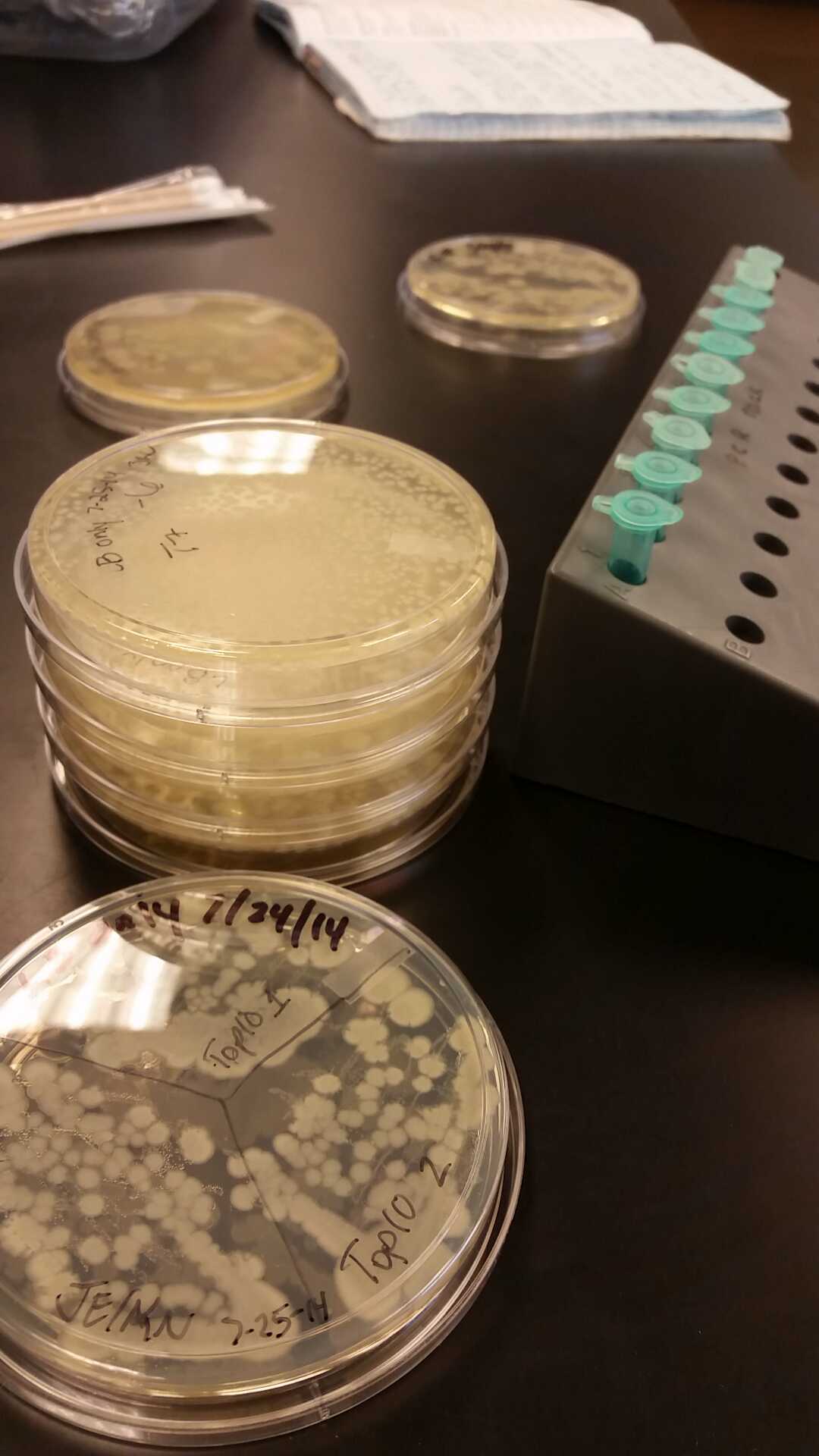
Week of 7/13/14 through 7/19/14
- This week we worked towards switching our part's backbone from pSB1AT3 to pSB1C3. We had some difficulty doing this. The first attempt was fruitless, but on our second attempt we had success and we now have a stock of our part on a chlor-resistant backbone.
- Unfortunately, We no longer have any stock of our original part. So, now we are again following the 3A Biolabs Biobrick Assembly Manual to reconstruct our parts K1477014 and K1477030. Our first try yielded nothing.
- Our second run through of the protocol resulted in the growth of red colonies on chlor plates after electroporation of our part into Top 10 cells. However, we need white colonies to grow because this will point towards the rfp being repressed when the three parts come together. Red colonies are a sign that we did not have a successful assembly of our parts.
Week of 7/6/14 through 7/12/14
- Last week Lyuda and Kevin ligated the composite part from the pSB1AT3, to the pSB1C3. We had difficulty getting the plasmid backbone to take the transformation. We then emailed the IGEM FAQ section and got an almost immediate response stating that the chlor backbone had a really low transformation efficiency. We tried again on 7-20 the ligation of both K1477030, and K1477014 with success but it takes like two days in the incubator.
- We are in the process of running a gel to prove our parts are the right size in base pairs to show proper replacement transformations.
- We have resuspended our composite parts in LB and the appropriate antibiotics to begin mass producing for concentration testing. We are awaiting the arrival of viruses for furthering our virus titering protocol. We now have duplicate lines of all composite parts growing and in suspension all prepared for moving into concentration testing. PROGRESS!!
- These are the first successful transformation plates of transforming the plasmid backbone from pSB1AT3 to pSB1C3 for the purpose of submitting to the IGEM registry.
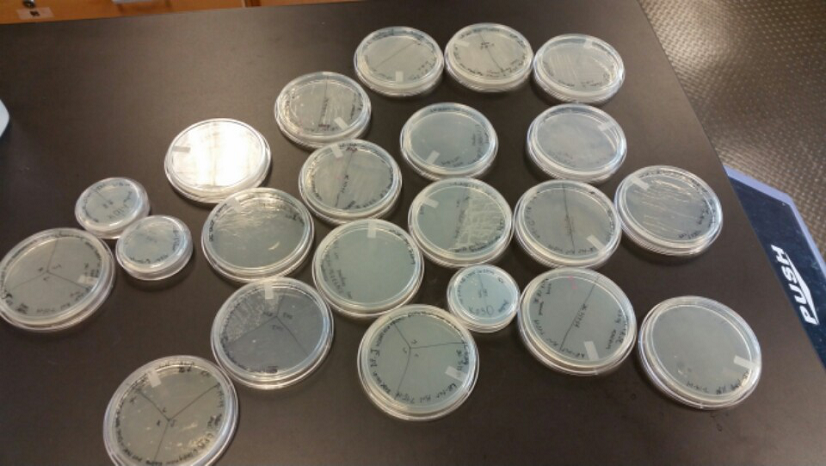
Week of 6/29/14 through 7/5/14

- Last week Lyuda and Kevin ligated the part K1477014 with J23102(constitutive promoter) I732020( LacZoperon Mutant) into pSB1AT3(destination plasmid).
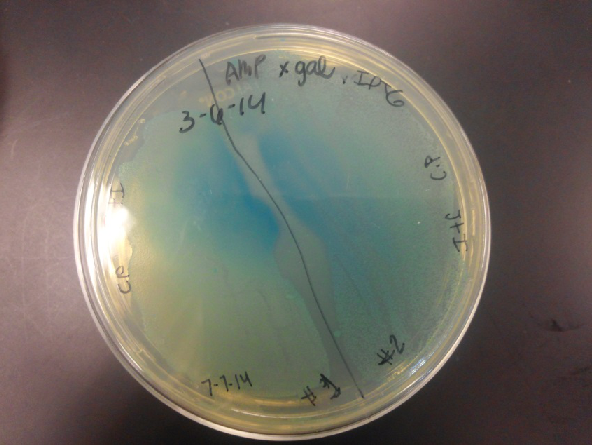
- The plates had a 1:7 ratio of white to red colonies. Transformants were selected from TET plates expanded in LB with the appropriate antibiotic, rolled in the 37 Celsius incubator overnight. The next day we plated on TET and 2 % X-gal plates. 2% X-gal plates were made by Kevin using the protocol in PT’s lab binder. X-gal plates contain galactopyranoside which detects the activity of converting lactose into glucose and galactase. Betagalactosidase activity is present in our K1477014 due to the indication of a blue color on the IPTG and X-gal plates.
- Ten 1.8 mL cryogenic vials with 50/50 of our cells from composite tube #1 and #2 and 40% glycerol were prepared and stored in the -80 freezer as reserve stock.
- Performed an upstream and downstream cut of I712074 (T7 promoter), K112806(Endolysin) with pSB1AT3(destination plasmid). Plated and picked colonies waiting to see results. We named the endolysin part as K1477030. Plan for this week is to finish the proposal, finish our Endolysin part and put the alpha generator onto a chloramphenicol plasmid backbone.
- Composite part (J23102 and I732020) on X-gal plates and regular ampicillin and tetracycline plates. The plates with no x-gal had white lawn growth with no red present.
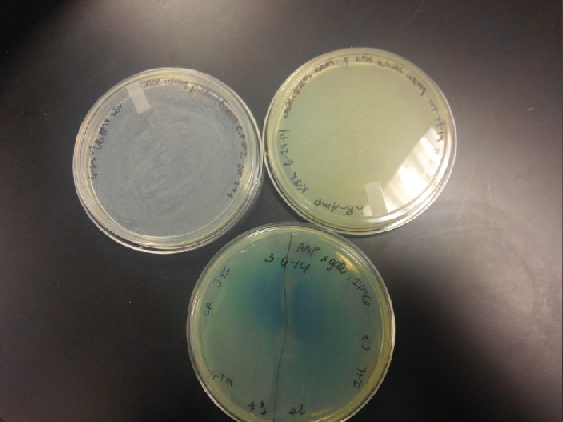
Week of 6/22/14 through 6/28/14
- Last week our team once again took the time to clean the IGEM box and autoclave everything inside the box including the pipettes. We were able to stop the contamination that seemed to be coming from the B.cereus. We are continuing to do everything in triplicates so our results would show replication and consistency.
- Our K1477014 part which was ligated with J23102 (p-con) and I732020 (LacZoperon Mutant) onto a destination plasmid (pSB1AT3) transformants were selected on TET, however when white colonies where picked from the tetracycline plate expanded with the appropriate antibiotic and LB, rolled for an hour, plated and put in incubator overnight, only a red lawn growth would appear every single time and not a single white colony present or visible. Our team came to the conclusion that during the first steps of the ligation process we made an error.
- We took all the resuspended tubes of J23102 and I73202 and purified by the miniprep procedure from a cell line derived from the electroporation of Top 10 cells on (05/25/14). We ran a gel to confirm the sizes of the DNA. Performed an upstream and downstream cut of the three parts. We plated the composite part, let it grow out and had good growth on all TET plates. Our purified DNA is stored in the glass front fridge on the left in a box.
- Both red and white colonies present on tetracycline plates; white colonies are a little bit smaller than the red
- Endolysin (K112806) and T7 promoter (I712074) tubes are purified, electroporated and gel ran on them. Upstream and downstream cut will be done on 7-2-14.
- Kevin performed the titer bacteriophage stock for the T7 virus (WARD’s Bacteriophage T7 2.5x10^7 pfu/ml) Virus grow in high concentrations, diluting them is a step that helps us count viruses effectively. To isolate bacteria I performed a dilution series of phage starting from 10^-1 through 10^-10 in LB broth. Kevin combined 4mL of 0.7% melted agar with competent Top 10 cells. Five tubes were labeled with soft agar. Soft agar was combined with the corresponding serial dilution and bacteria and poured on a warm agar plate.
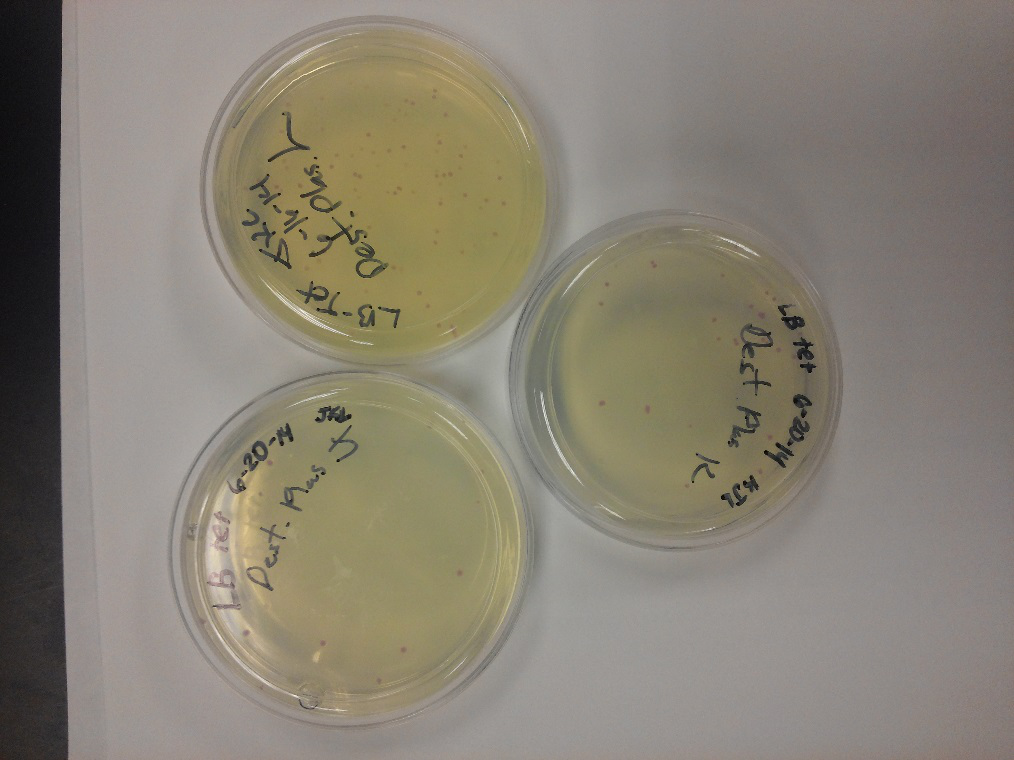
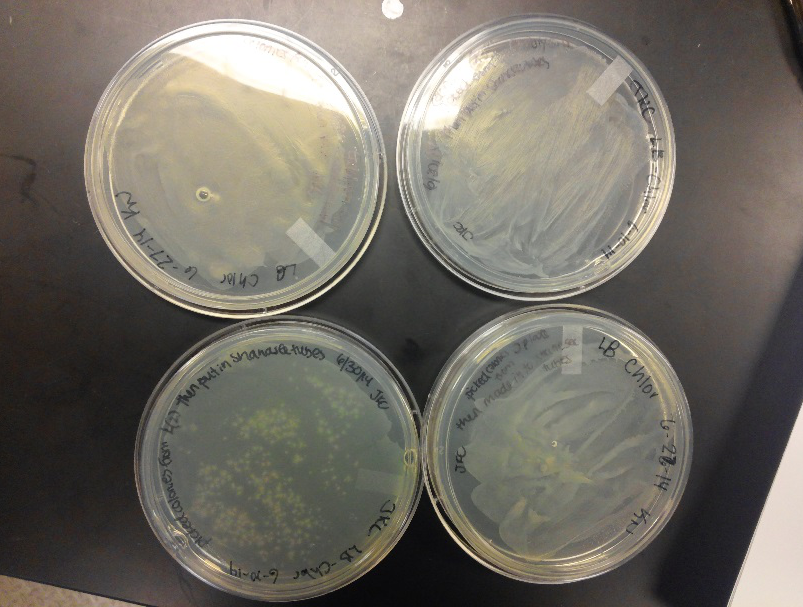
Week of 6/15/14 through 6/21/14
- Progress of our three man effort. We took a little time this week to clean out our IGEM box due to a contamination that seems to be spreading! We had a meeting among us to re-evaluate our plan. Decided that we would continue everything in triplicate so our results would show replication.
- We began the week by ligating J23102 (p-con) with I732020 (LacZoperon Mutant) onto a destination plasmid (pSB1AT3). Since we had various registry DNA parts of these individual cells, we decided to use different parts for each of us. We all used the same J23102, two different I732020 lines, and all three used a different pSB1AT3 line. Apparently Lyuda’s line of pSB1AT3 was not viable as her Destination Plasmid did not grow on Tet plates. The other two of us had plenty of growth with large red colonies and small white colonies, indicating that some of the J23102 must have went backwards to the original J23102 with red cells growing on Amp.
- We then resuspended three lines of J23102, three lines of white chlor cells I732020, and three lines of our K1477014 (The new number for Ivy Tech South Bend). We also pulled a new line of k112806 (endolysin) from the 2014 registry plate #3, well 12K. We then electroporated and plated this line in triplicate, we also started three lines of I712074 (T7 promoter.) With this we will transform the promoter, k112806 ,and LacZoperon with the Ω part into destination plasmid pSB1AT3 to get our second registry part to submit.
- We also made new plates, Amp large plates and all four antibiotic resistances on the small plates. We then tested both controls on all four antibiotics. Amp plate amp seems to grow. B. cereus has the capability to adapt to antibiotics such as chlor.
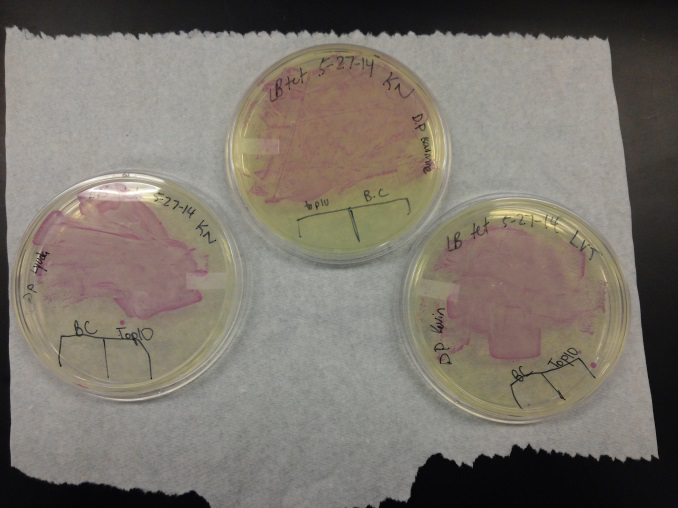
Week of 5/25/14 through 5/31/14
- We transformed Top 10 cells through electroporation with registry parts K112806 (Endolysin,) I732020 (LacZoperon Mutant,) J23102 (p-con) and I712074 (T7 promoter.)
- After streaking on plates, and leaving them in the incubator for a day, there was growth on all plates, showing that a successful transformation occured.
- Plates containing J23102 were red-another sign of success. Unfortunately, it looks like contamination has arisen as well, and is spreading on plates with I712074 and K112806 growth.
- The contamination persisted all week, but we were able to get plates with transformed parts that contained no contamination growth.
- We then ran a DNA purification protocol of J23102 and I732020 to get them ready for a biobrick assembly protocol.