Interlab Study
Expectation
J23101.E0240 and I20260 are two Biobricks consisting of the same insert. The insert consists of the promoter J23101, the RBS B0032, the GFP coding sequence E0040 and the Terminator B0015. The only difference between these Biobricks is the vector backbone, pSB3K3 for I20260 and pSB1C3 for J23101.E0240 respectively. pSB1C3 is a high copy plasmid while pSB3K3 is low to mid copy plasmid. Therefore a higher fluorescence is expected of J23101.E0240 compared to I20260 even though they share the same insert.
J23115.E0240 differs from J23101.E0240 only in the promoter. The J23115 promoter is a lot weaker (the mutated version K823012 was used) compared to J23101. Therefore, a lot lower fluorescence is expected, if any.
B0015 was used as our negative control as the insert only contains a terminator and no expression cassette for GFPmut3b, therefore no fluorescence was expected.
Results
After observing the Optical density (OD) and fluorescence for 18 hours whilst taking samples every 2 hours, the following results were obtained:
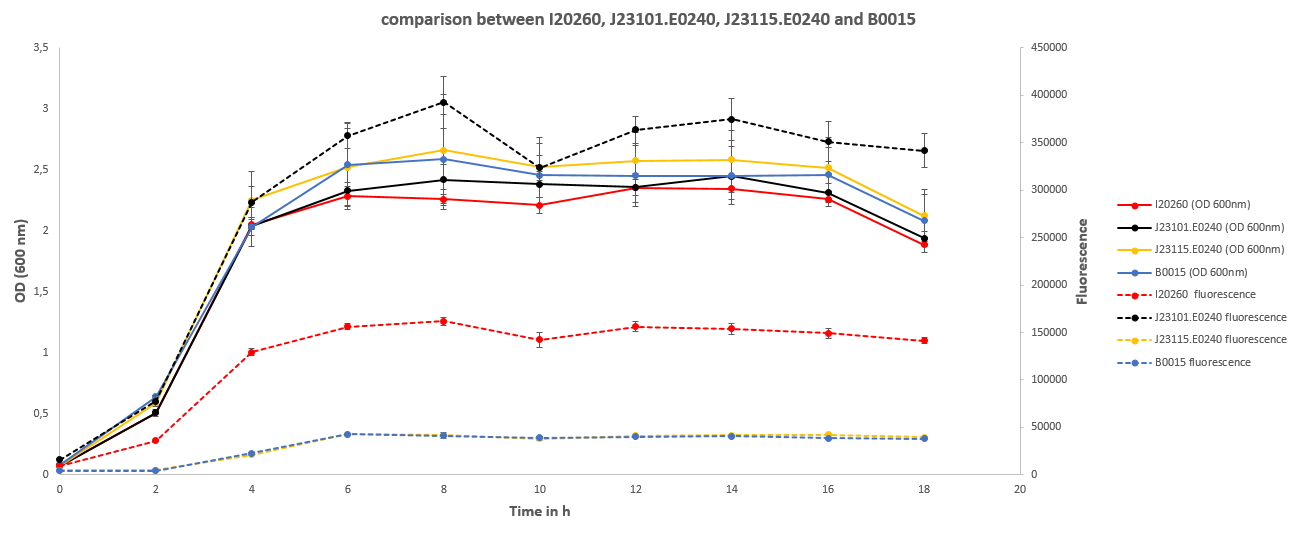
This shows that all cultures had the same Optical densities throughout the experiment. After the exponential growth phase the statinary phase started shortly after 4 hours. The OD didn't change from thereon until the after 16 hours were it started to decline again.
The fluorescence followed largely the pattern of the OD, but differed a lot in between the different cultures. J23101.E0240 showed about 3 times the fluorescence of I20260 and about 10 times the fluorescence of B0015 and J23115.E0240 which both showed no difference between each other.
Discussion
The OD is an indirect measure of the biomass in the shake flask. Through the correlation of both measurements the results show that the biomass of all cultures showed no significant enough differences to affect the fluorescence data. Therefore the fluorescence data can be interpreted as direct result of the fluorescence per cell instead of a difference in the amount of fluorescing cells.
The fluorescence data therefore shows a strong difference between the I20260 and J23110.E240. Even though both inserts are the same, the difference in fluorescence is as expected different because of the difference of the plasmid backbone. The high copy plasmid pSB1C3 shows a 3 times higher fluorescence per cell then the low - to mid copy plasmid pSB3K3. This can be directly related to the number of plasmid in the cells coding for GFP.
J23115.E0240 and B0015 show both no significant fluorescence. The increase for the 4 hours is explained by the increase in OD and therefore noise in the media. B0015 behaves therefore as expected.
J23115.E0240 in its original, non mutated state was supposed to show a slight, even though less fluorescence then J23101.E0240. But the mutatinons introduced made the promoter non functional which lead to no expression of GFP and therefore no observed fluorescence.
Materials and Methods
Constructs and strains
All constructs used were transformed into NEB 10 Beta cells. The constructs I20260 as well as B0015 were taken directly from the iGEM 2014 distribution plates and transformed directly. The constructs J23101.E0240 as well as J23115.E0240 were made using the 3A Assembly. Therefore the subparts J23101, J23115 as well as E0240 were transformed directly from the 2014 distribution plates into NEB 10 Beta cells. Afterwards the plasmids were recovered using the illustra plasmidPrep Mini Spin Kit. Afterwards the purified plasmids J23101 and J23115 were cut with the restriction enzymes EcoRI and SpeI, while E0240 was cut with XbaI and PstI. The restricted plasmids were then ligated together using the T4 DNA Ligase. Afterwards the Ligation product was ligated into the pSB1C3 linearized backbone provided by iGEM headquarters with the 2014 distribution after being cut with EcoRI and PstI (All restrictions and ligations were performed using enzymes and buffer of the NEB iGEM Kit). The final product was once again transformed into NEB 10 Beta cells.
The sequences of the resulting construct were confirmed by sequencing.
The sequencing data (consensus sequences) can be found here.
Note: We used the mutated version of J23115 as sent out by the iGEM headquarters. The mutation makes J23115 effectively the same promoter as K823012. We will still refer to the Promoter as J23115 though to keep it more easily recognizable with the other Interlab study results.
Inoculation and Cultivation
The cultivation of our cultures was performed in 50 ml LB medium in 500 ml shake flasks at 37 degrees Celsius and 300 rpm shaking frequency. Appropiate antibiotics were added to each media (Kanamycin for I20260, Chloramphenicol for B0015, J23101.E0240 and J23115.E0240). Both antibiotics were added from a 1000* stock stored at -20 degrees Celsius for a final concentration of 35µg/ml Chloramphenicol and 50µg/ml for Kanamycin respectively.
The precultures were inoculated from the same cryo stock every time under a sterile clean bench. They were cultivated for 16 hours and then sampled for OD measurement with a spectrophotometer. Then 2 ml of each preculture were centrifuged (5 minutes, 6000 g) and then washed twice with PBS buffer. Afterwards, all cultures were inoculated to have the same starting OD, once again done under sterile conditions inside a clean bench.
Sampling
To draw samples from the shake flask, the shake flasks were taken out of the 37 degrees Celsius room and brought onto a nearby bench. There 3 ml of sample were taken out next to a Bunsenburner flame and put into 2 ml cuvettes. As soon as all samples from all flasks were taken the flasks were taken back inside the 37 degrees Celsius warm room and shaking was restarted.
The whole process of taking samples for all 12 flasks (3 biological replicates for each construct) took 5 minutes and samples were taken every 2 hours.
After 4 hours we had to dilute the sample to a concentration of 20 % and from the 6th to 18th hour we had to dilute to a concentration of 10 %. Dilution occured in LB medium.
After the measurement of OD in the spectrometer 100 µl of each sample were taken out and put on a 96 well plate (Thermo microfluor 1, flat-bottom, black) to measure fluorescence. Each measurement occured in a technical triplicate, resulting in 36 different samples being measured for every sampling step.
Measurement of OD using a Spektrophotometer
For OD measurement the Unico Spectrophotometer 1201 of Fisher Bioblock Scientific was used. OD measurement was taken at 600 nm and we used pure LB medium (from the same batch as the medium used for cultivation) as our blank. We only measured OD up to 0,8. At a higher OD we diluted the sample with LB medium (again from the same batch as our cultivation medium). Dilution was done by presetting the LB medium into cuvettes, then adding our cultivation sample and vortexing it thoroughly. The solution was allowed to settle before measurement.
Measurement of fluorescence using a Microplate reader
Measurement of fluorescence was performed using the Synergy Mx from BioTek with the Gen5 software.
Measurement was taken using the following parameters:
| Parameter | value
|
| Software version | 2.1.2014
|
| Reader Type | Synergy Mx
|
| Read | GFP 100
|
| Measurement | fluorescence endpoint
|
| Measurement range | full plate
|
| Filter | filter set 1
|
| Excitation | 496/9,0
|
| Emission | 516/9,0
|
| Gain | 100
|
| Read Speed | normal
|
| Delay | 100 msec
|
| Measurement s/data point | 10
|
| Read height | 8 mm
|
We used LB medium as our blank. Following from our sampling technique we used the same dilutions as for the measurement of OD.
|
 "
"
