 "
"
Team:SUSTC-Shenzhen/Notebook/CRISPR/5*UAS-plasmids-Fill-in
From 2014.igem.org
Notebook
Elements of the endeavor.
Contents |
5*UAS plasmids Fill-in
2014/8/6 To eliminate the EcoRI site on the 5*UAS plasmidMaterials
- dNTPs
- Enzymes&Buffer:
- EcoRI, Cutsmart Buffer
- T4 DNA polymerase, T4 DNA polymerase Buffer
- T4 DNA ligase, T4 DNA ligase buffer
- Kit:
- TIANgel Purification Kit
Methods
Restriction digestion for 5*UAS plasmid with EcoRI
| Total volume | Buffer | DNA | EcoRI | ddH2O |
|---|---|---|---|---|
| 20μl | 2μl | 8μl | 1μl | 9μl |
Start time: 9:41am End time: 2:00pm
Electrophoresis & Agrose gel DNA extraction to remove enzymes and competent of Buffer
Loading system:
| Total volume/well | DNA | Dye | TAE |
|---|---|---|---|
| 24μl | 5μl | 4μl | 15μl |
Running conditions:110V , 30mim DNA extraction:
Performed as the protocol provided by TIANgel Purification Kit, except that we eluted the DNA sample twice at the last step (as we always done before).
Concentration of the purified DNA fragments:
Poly A: 24.8ng/ul G-PB5: 42.2ng/ul
Do 5’-end fill-in with T4 DNA polymerase
Protocol
- DNA should be dissolved in 1X NEBuffer 1-4 or T4 DNA Ligase Reaction Buffer supplemented with 100 µM each dNTP.
- Add 1 unit T4 DNA Polymerase per microgram DNA and incubate 15 minutes at 12°C.
- Stop reaction by adding EDTA to a final concentration of 10 mM and heating to 75°C for 20 minutes .
- CAUTION: Elevated temperatures, excessive amounts of enzyme, failure to supplement with dNTPs or long reaction times will result in recessed ends due to the 3´→ 5´ exonuclease activity of the enzyme.
Reaction system:
| Poly A | G-PB5 | |
|---|---|---|
| DNA | 17.8ul(441.44ng) | 17.8ul(751.16ng) |
| 10X T4 DNA Ligase Reaction Buffer | 2ul | 2ul |
| dNTP | 0.2ul | 0.2ul |
| T4 polymerase (3U/ul) | 0.2ul (Theoretical value: 0.1471ul) | 0.3ul (Theoretical value: 0.2503ul) |
| Total volume | 20.3ul | 20.2ul |
EDTA
We prepared 100mM EDTA stock solution. To stop the reaction, we added 1ul EDTA (100mM) to 19ul reaction liquid. Heat 75°C, 20min Blunt end ligation with T4 DNA ligase
- Till now, the concentration of DNA
- Poly A reaction system:21.0ng/ul
- G-PB5 reaction system:35.3ng/ul
- ligation system:
| DNA | 10X buffer | T4 ligase | ddH2O | |
|---|---|---|---|---|
| Theoretical volume | 37.5ng | 2ul | 1ul | Add to 20ul |
| Poly A(1,2) | 2ul | 2ul | 1ul | 15ul |
| G-PB5(1,2) | 1ul | 2ul | 1ul | 16ul |
- Protocol:
- Incubate 16°C, overnight (16h)
- Heat inactivation, 65°C, 10min
- Chill on ice
- Remove EDTA:
- we add 1ul pfu (with MgSO4) buffer to each ligation system (totally, 4*tubes: polyA1, polyA2, G1, G2), in order to neutralize the inhibit effect of EDTA.
- Restriction digestion using EcoRI to re-linearize those sticky end ligations, thus selecting out those successful ones.
- Till now, the concentration of DNA
- Poly A reaction system:37.5ng/20ul=1.8ng/ul
- G-PB5 reaction system:37.5ng/20ul=1.8ng/ul
- Digestion system:
- Till now, the concentration of DNA
| Total volume | EcoRI (20U/ul) | DNA | 10X NEBuffer | ddH2O | |
|---|---|---|---|---|---|
| Theoretical volume | 10ul | 1 unit | X ul (0.1ug) | 1ul | Add to 10ul |
| Poly A1 & G-PB51 | 22.1ul | 0.1ul (2 unit, obviously we have excessive enzyme) | 20ul(about 37.5ng) | 2ul | - |
Also, design a non-digestion system as control
Poly A2 & G-PB52 didn’t undergo restriction enzyme digestion.
Transformation
Performed as the usual protocol
- 5ul ligation reaction to 50ul competent cell.
- Incubate: 2014.8.7 9:30pm~2014.8.8 2:30pm (17h)
Results
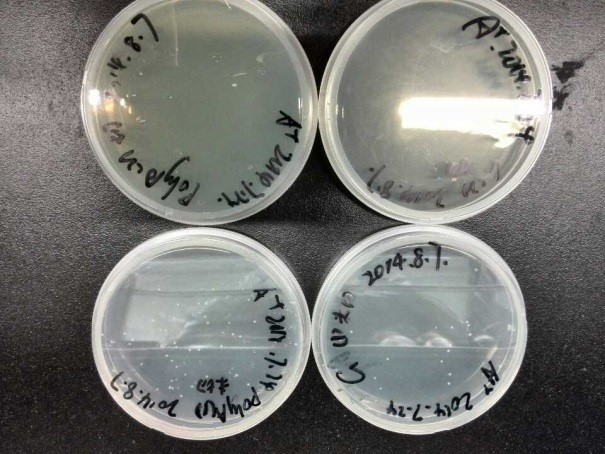
- From the picture, we can see that, there were no colonies growing on the plate that were grown with plasmid-digested bacteria(Poly A1, G1). While for those bacteria which were transformed with undigested plasmid (the control group: Poly A2, G2), several colonies have already appeared on the agar plate.
- This meant the successfully transformed colonies were all false positive, of which the plasmids were all formed by sticky-end self-ligation, rather than blunt ends which should be generated via 5’ end Fill-in.
- Finally, in a word, our 5*UAS plasmids Fill-in has failed.
Points that can be improved in the next time:
- Do not undergo Agrose gel DNA extraction after the-first-time enzyme digestion, since the buffer for digestion is also suitable to T4 polymerase. Thus, a relatively high concentration of DNA can be assured.
- After the polyreaction step of Fill-in, recover DNA by agarose gel DNA extraction, to remove EDTA, buffer etc. most of which might affect the following experiment.
- Design a concentration gradient when performing the T4 ligation step (30ng, 100ng, 200ng DNA). Make sure that there is always a DNA concentration involved in the optimum range for ligation.