 "
"
Team:Heidelberg/Project/Reconstitution
From 2014.igem.org
– Bringing Parts together
ABSTRACT
In our intein toolbox we provide fusion and activation as principle adaptable for nearly all proteins that exhibit a modular structure. To prove this principle we split superfold GFP in two halves at amino acid position 64/65 and joined each to the corresponding split intein NpuDnaE. In this experiment we successfully reassemble the protein halves and restored fluorescence of sfGFP. The fluorescence was measured via TECAN plate reader and FACS analysis, as well as visualized on the Western Blot. Split fluorescent proteins provide an easy to use read-out for the efficiency of intein splicing and can be further used for large screenings as utilized in our LOV-mutant screen
Contents |
Introduction
A common strategy for investigating molecular and cellular biological questions is the use of fusion proteins and to control the functions of these proteins in a spatial and temporal manner. Instead of manipulating proteins on the genomic level, we aimed at editing proteins post-translationally. By using our intein toolbox, one is able to fuse proteins and/or protein tags , as well as reconstitute the function by complementing two formerly split halves of a single protein, and thereby restore the function of the protein. Mechanistically, the reconstitution of split proteins is identical with the fusion of different proteins to their tags.
To demonstrate the restoration of function of a formerly split protein, we choose a set of fluorescent proteins, whose function can, when properly reassembled, easily be read out using their florescence. Split fluorescent proteins are rarely used in the context of intein splicing. However, they are widely applied in bimolecular fluorescence complementation (BiFC) assays [1]. This approach is based on the complementation between fragments of fluorescent proteins that reconstitute its fluorescence when brought into proximity by associated interacting proteins.
Experimental Procedures
Selection of Split Sites
It is important to select the correct site to split fluorescent proteins, so that the reconstitution of the fluorescent properties is possible. Therefore, we selected split sites according to previous published research, where the splice sites are mostly located in the flexible parts connecting the β - barrels of the fluorophore. Additionally to fluorescent proteins we choose to create two versions of split luciferases. A list of all designed constructs is depicted in the following table:
| Protein | Split site | Comment |
|---|---|---|
| mRFP | 154/155 | Split between β - barrel 7 and 8 [2] |
| mCherry | 168/169 | Split between β - barrel 8 and 9 [3] |
| GFP | 157/158 | Split between barrel β - 7 and 8 [4] |
| sfGFP | 64/65 | In front of the chromophore region [5] |
| Firefly Luciferase | 437/438 | In flexible tether between the two subunits [6] |
| Renilla Luciferase | 229/230 | split between barrel β - 7 and 8 [7] |
Originally we wanted to evaluate the reconstitution of all created constructs by screening the signal to noise ratio using FACS. Noise, or background, can occur due to the possibility of reconstitution of two seperated halves without a splicing reaction, a feature used in the BiFCs assays.
Unfortunately, for time reasons we could focus solely on the reconstitution of superfolder GFP (sfGFP) and mRFP. We chose sfGFP to prove the principle of activation by the restoration of the protein halves and to characterize the splicing reaction of the NpuDnaE intein via FACS analysis and Western Blot technique.
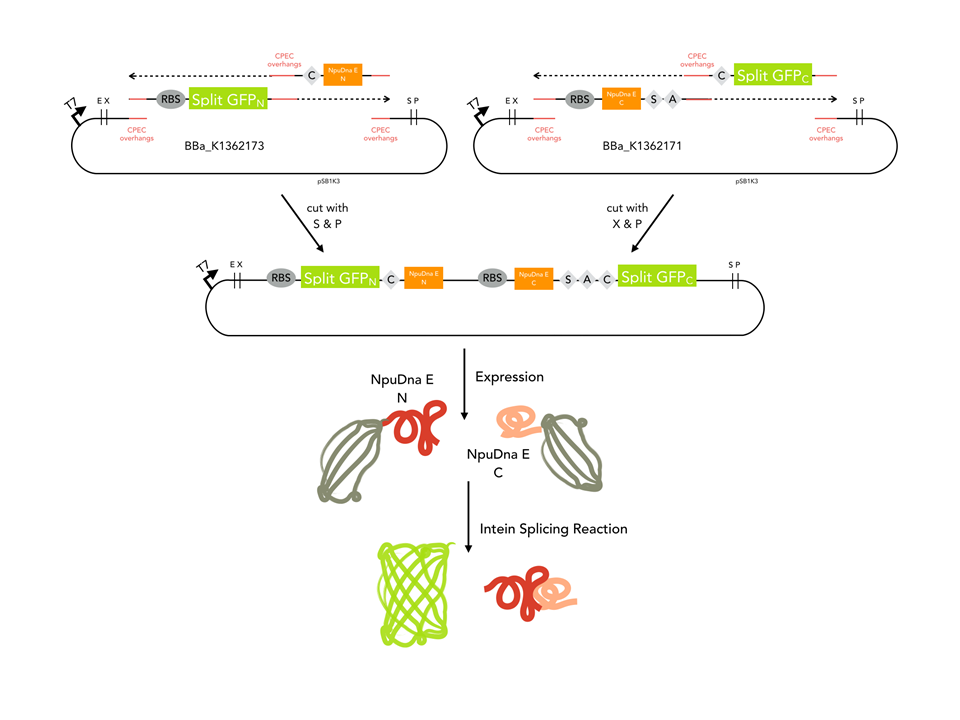
GFP_N was amplified from BBa_K1362173, CPEC overhangs and the NpuDnaE_N were in the PCR primer. NpuDnaE_C was amplified from BBa_K1362171 with Oligos beinhalten CPEC overhangs and the GFP_C. Both PCR products were cloned in a biscistronic expression backbone using CPEC.
Cloning strategy
Since our assembly construct with two insertion sites was not ready at this point, we chose to follow a fast track cloning strategy to initiate trans - splicing of split sfGFP. Therefore, we cloned the N-terminal protein part in front of the N - partial split intein NpuDnaE and the C-terminal GFP part (65-225) downstream of the C- portion of the intein using CPEC. The N- and C-terminal parts of GFP were cloned in a bicistronic expression backbone to allow expression from the same promoter via standard biobrick cloning. Also, non-splicing variants were generated by changing the essential cysteine at the N-terminal part of the N-intein to glycine (C > G) and the serine and asparagine in the C-intein to alanine and glycine (SA > AG). In order to splice, the inteins need a cysteine at the amino acid position where the splicing reaction will occur. In this case, the splicing product will harbour a cysteine at amino acid position 65. To ensure that the inserted cystein does not interfere with the fluorescence of the reconstituted sfGFP, we cloned a positive control of sfGFP by insertion of the cystein using mutagenesis PCR.
Fluorescence measurement
We expressed the split sfGFP construct with the various controls in BL21(DE3). 5ml cultures were inoculated with 500 µl of an overnight culture and induced with 1mM as soon as the cultures reached the OD600 of 0.8. The cultures were incubated at 37 °C for further 4 hours. Subsequently, the fluorescence intensity was measured in a TECAN plate reader or in a flow cytometer. To ascertain the best excitation and emission wavelength in the plate reader we tested several excitation and emission wavelengths. Excitation at 475 nm and emission at 512 nm turned out to be most suitable for sfGFP. To validate our results we conducted several assays with a series of biological replicates following the same experimental layout (Figure 2). 300 µl of each sample were taken to measure the fluorescence in the plate reader on a black 96 well plate and a 1:1000 dilution of the original culture was prepared for the FACS measurements. The laser settings were adjusted to 488 nm excitation and 497 - 522 nm emission range. The samples were loaded as described in the illustration below (Figure 2). Besides the in vivo expression and splicing reaction, an in vitro reaction mix of N- and C-terminal split GFP-NpuDnaE was set up. In order to conduct a Western Blot the cells were harvested by centrifugation for 5 min at 3270 rcf at 4 °C. Subsequently, they were resuspended in 500 µl PBS on ice and sonicated for 2 min at 50% power on ice. The samples were centrifuged at 18000 rcf for 10 min and the supernatant was resuspended in 500 µl PBS on ice. For the Western Blot and Coomassie Gels, samples were mixed with 100 µl 5x Lämmli-Buffer and 10 µl were loaded on the SDS-Gel.
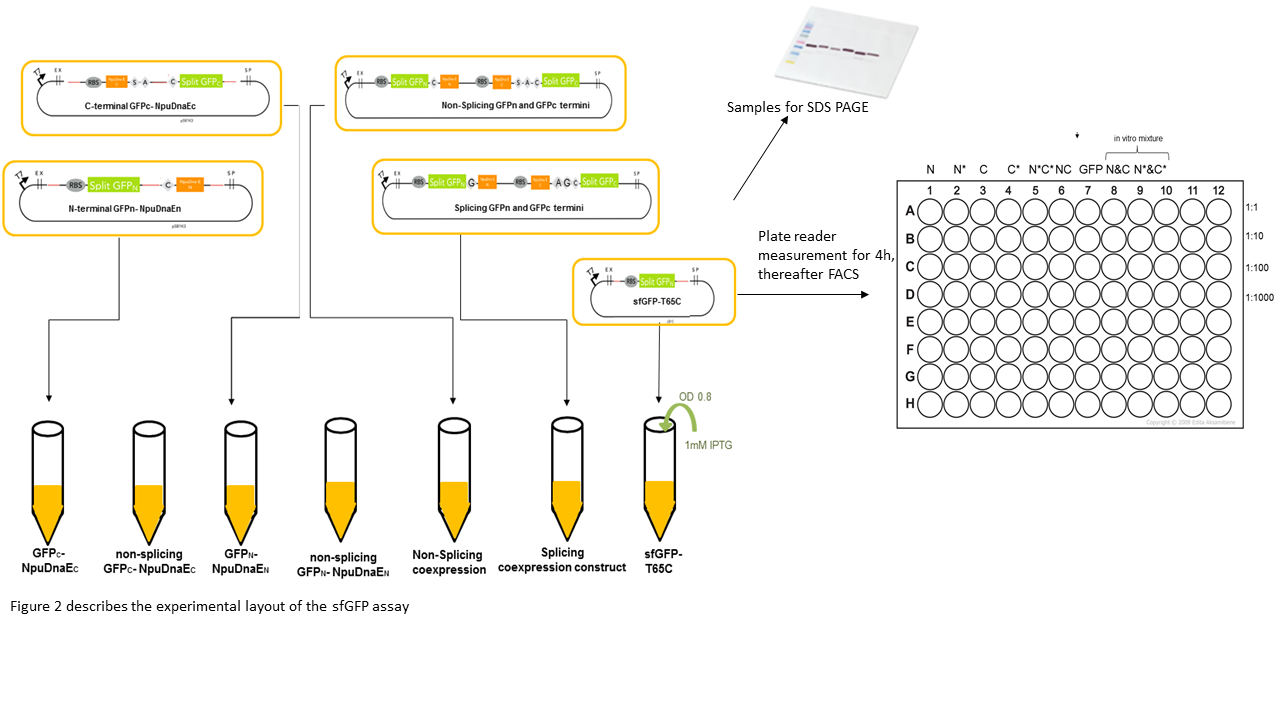
Cultures were grown to OD 0.8, followed by 4 hours of induction with 1mM IPTG. Samples were subsequently analyzed by SDS Gels or by measuring the fluorescence by plate reader or FACS.
Results
To proof our idea of reactivating protein function by using inteins to fuse split parts, we decided to use fluorescent proteins. Fluorescent proteins are widely used, and the read out is simple. After expression of the sfGFP halves and split inteins, either as separated N- and C- terminal constructs or combined on one plasmid, the fluorescence was meassured via plate reader and FACS.
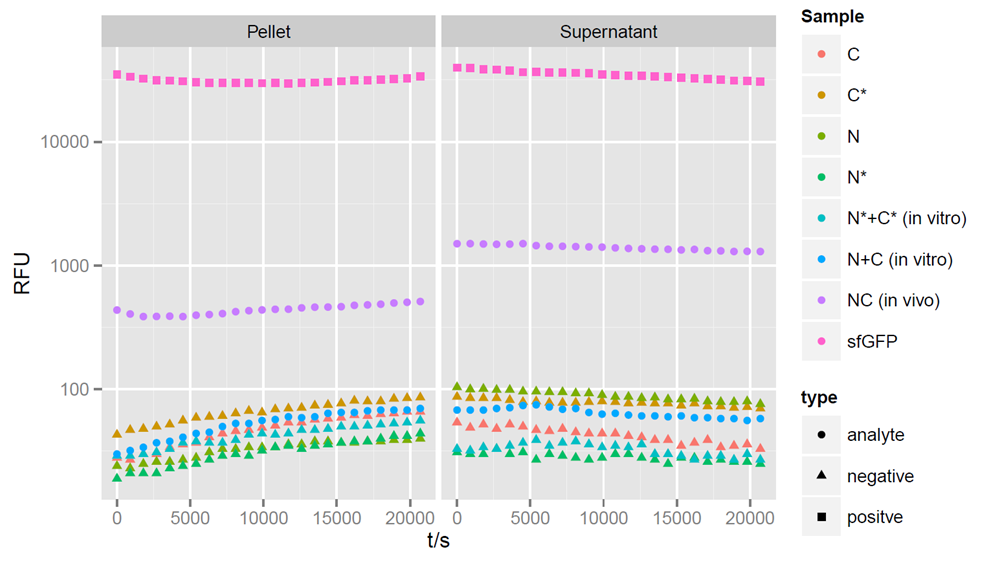
Fluorecence intensities detected at 475nm exitation and 512 nm emission wavelength for a period of 6 hours after induction. Split halves and splicing controls show no fluorescence. Simultaneous expression of the split parts leads to a strong increase of sfGFP fluorescence.
After 4 hours of expression, a clear fluorescent signal is observed within in vivo samples with both, the N - and C - terminal parts of sfGFP, showing that fluorescence can be restored by trans-splicing (Figure 3). Unfortunately, fluorescence can not be observed in in vitro samples, indicating that no splicing reaction took place. The in vivo fluorescent signal is stronger in the supernatant than in the pellet. However, it is not as strong as the sfGFP control. The fluorescent signal of reconstituted sfGFP is about 100-fold stronger then the negative controls, when only a single part of sfGFP, or a non-splicing variant is expressed. The mixture of N- and C- terminal construct in vitro after lysis of the cells revealed no fluorescence, indicating that no splicing reaction took place.
However we were missing an essential control in this experiment. In this experiment, we could not show the in vivo fluorescent signal when both non-splicing parts of sfGFP are expressed. This control is required, since there is the possibility that both parts of the protein come in close proximity to each other and form the complete protein independently of a splicing event.
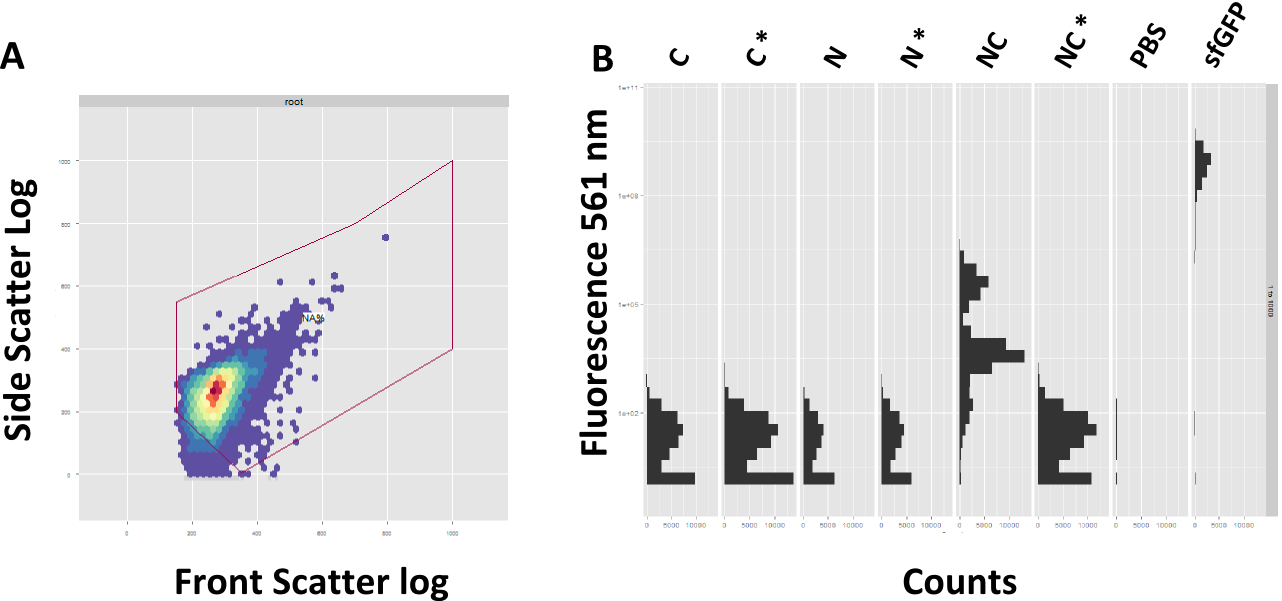
A: Exemplary Gating of the sample. Front Scatter depicts the size, Site Scatter Granualarity of each counted event. B: Successful reconstituion of sfGFP after 4 h
In course of the experiments we focused on the read-out via FACS, since this approach promises a more precise evaluation of fluorescence in single cell resolution. Prior to each FACS experiment, the machine had to be adjusted. On one hand, the gain was adjusted to the strongest fluorescent sample, the positive control, and the correct subpopulation of events had to be selected, a process termed gating. The front scatter depicts the size of the event, the side scatter granualarity. A proper gating is mandatory to assure correct measuring of single bacteria, instead of aggregated bacteria or debris (Figure 4A).
The FACS data is showing a clear spectral shift towards the green fluorescence range for the splicing construct compared to the non-splicing control (Figure 4B). Likewise the single halves do not show fluorescence on their own. However, after 4 hours of induction the splicing product does not reach the same amount of fluorescence as the sfGFP.
Since the fluorescent signal in both, the plate reader and the FACS, can be misleading, due to the possible reconstitution without splicing, we decided to verify our results by Western Blot. Prior to blotting, the proteins are denaturated. Thus, non-spliced sfGFP would appear as two separate parts on the blot, whereas irreversibly spliced sfGFP would appear as a single band. We could detect the single sfGFP parts, as well as the reconstituted sfGFP by the attached HIS - Tag.
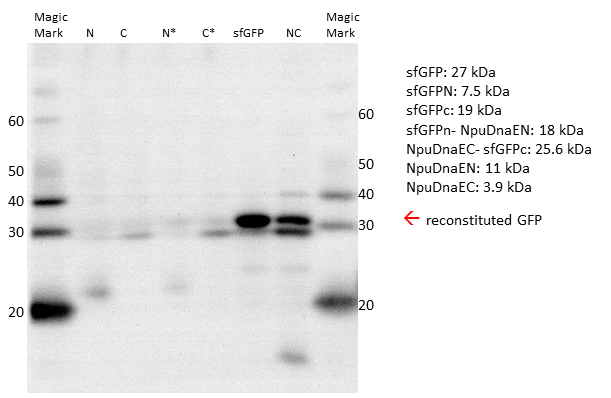
Western Blot with His-antibody. Single intein parts and the non-splicing controls are not sufficient to form a reconstituted sfGFP. Only expression of both parts leads to restoration of sfGFP.
The Western Blot of the expressed constructs shows a significant amount of sfGFP reconstitution. Neither of the split intein halves reconstitutes sfGFP, only when both intein halves are expressed there is a sfGFP signal. Upon reconstitution, the splicing product is forming, which is only visible when restoration occurs. Interestingly, while there is still a strong band at the size of the NpuDnaEc-sfGFPc (C), there is no sfGFPN-NpuDnaEN (N) observable, indicating that the N - part is the limiting factor in the splicing reation. Considering the previous experiments, we assume that there is only an irreversible reconstitution when both, the N - and C - part are expressed, and that the meassured fluorescence is based on the splicing reaction.

Time course of the reconstitution process. sfGFP is restored fast and the level of sfGFP remains constant. Please note that the order of the samples is different in panel A and panel B
Next, we wanted to analyze how fast the reaction of the two parts occurs (Figure 6). Therefore, we induced cultures for a set of timepoints, ranging from 2 to 10 hours and analyzed the amount of reconstituted sfGFP by FACS. sfGFP is reconstituted fast, and remains fluorescent over the whole time period, whereas no fluorescence was detected in the controls.
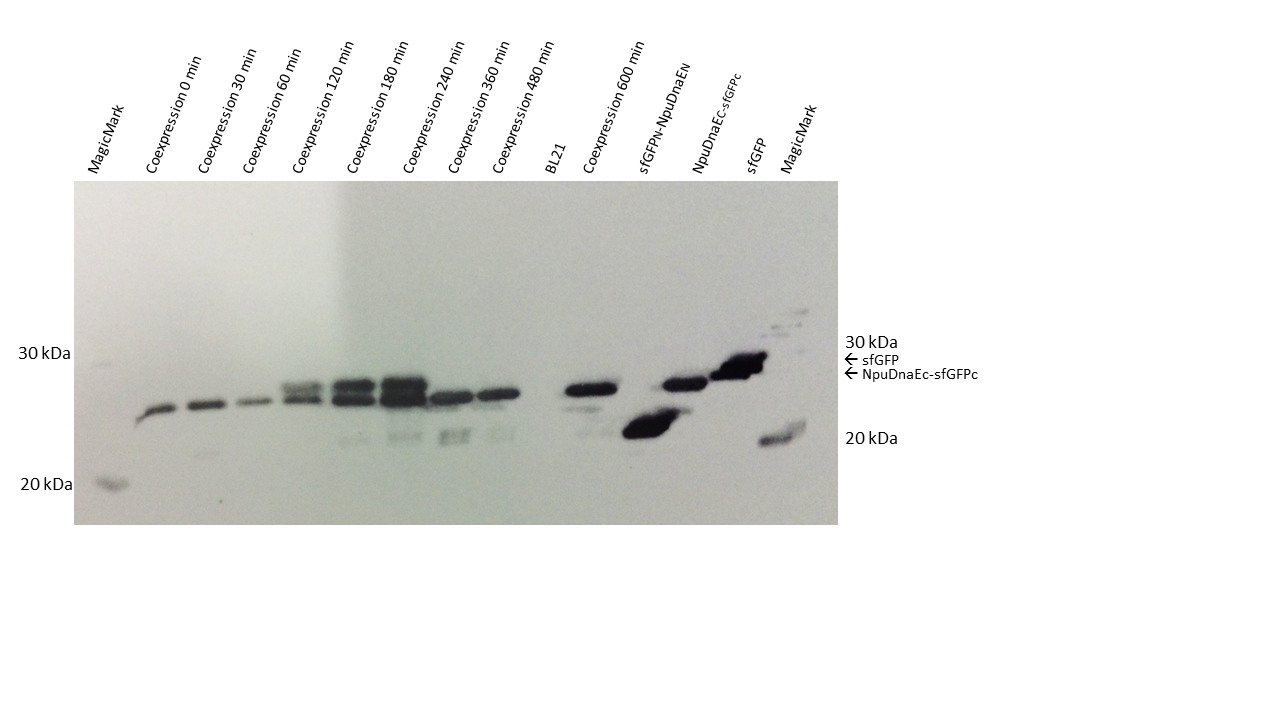
Time course of the reconstitution process. sfGFP is restored after 2 hours, the maximum is reached after 4 hours.
Additionaly to the FACS assay, we wanted to assure irreversible splicing and blotted protein samples from the same cultures. In contrast to the FACS data, the sfGFP signal is visible after 2 hours. The splicing product increases between two and four hours after IPTG induction. Unfortunately, there is no reconstituted protein after 6 hours or at any posterior time point. One reason might be that the samples were mixed up, or that degradation of the reconstituted protein occured, even though this is unlikely since both GFP - parts are continously produced during induction.
Discussion
Using FACS and plate reader data we were able to demonstrate reconstitution of fluorescence by NpuDnaE intein trans- splicing. The fluorescence increases significantly above the background that is produced by the non-splicing co-expressed control, and no reconstitued sfGFP is dectable upon denaturation.
The split sides within the protein seems to be very important for this approach and the suggested split region for the other fluorescent proteins and luciferases still needs to be tested in the context of intein splicing.
It is not clear, why the C-terminal NpuDnaEc-sfGFPc construct is always expressed at a higher level compared to the N-terminal construct, although both share one promoter (bicistronic expression) and occupy identical ribosomal binding sides. Usually, the N - terminal construct is expressed slightly higher. This is probably the cause why the splicing reaction seems incomplete on the Western Blot. Since there is an excess of the C-terminal portion the amount of splicing reaction is determined by the access of N-terminal construct. This could be avoided by adjusting the expression levels using distinct promoters and ribosomal binding sides. Another crucial question is the missing splicing reaction after 6 hours of IPTG induction in the time series of sfGFP reconstitution. It still needs to be clarified, if this is due to mistakes by the experimenter or attributed to biological causes. The bands that do not show the splicing product exhibit a higher amount of sfGFPN-NpuDnaEN, indicating that the splicing reaction itself probably does not take place. Since the same samples were used in the FACS measurement, it is likely that there was a miistake during the preparation of the sample for the Western Blot. The reconstitution of fluorescent proteins resembles a nice approach to prove the principle of fusion and activation of proteins by intein splicing. The split fluorescent proteins reveal an easy read-out, which can be beneficial for large screening applications. In our attempt to control intein splicing by light, we used this approach to screen for inLOV - constructs.
References
[1] Hu, C-D, & T.,K., Kerppola. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 21(5), 539-545 (2003).
[2] Jach, G., Pesch, M., Richter, K., Frings, S., & Uhrig, J., F. An improved mRFP1 adds red to bimolecular fluorescence complementation. Nature Methods, 3, 597-600 (2006).
[3] Furman, J., L., Badran, A., H., Shen, S., Stains, C., I., Hannallah, J., Segal, D., J., Ghosh, I. Systematic evaluation of split-fluorescent proteins for the direct detection of native and methylated DNA. Bioorg Med Chem Lett, 19(14), 3748-3751 (2009).
[4] Oyawa, T., Takeuchi, M., Kaihara, A., Sato, M., Umezawa, Y. Protein splicing-based reconstitution of split green fluorescent protein for monitoring protein-protein interactions in bacteria: improved sensitivity and reduced screening time. Anal. Chem, 73, 5866-5874 (2001).
[5] Aranko, A., S., Oeemig, J., S., Kajander, T., & Iwai, H. Intermolecular domain swapping induces intein-mediated protein alternative splicing. Nat. Chem. Bio., 9, 616-622 (2013).
[6] Ozawa, T., Kaihara, A., Sato, M., Tachihara, K., Umezawa, Y. Split luciferase as an optical probe for detecting protein- protein interactions in mammalian cells based on protein splicing. Anal. Chem, 73, 2516-2521 (2001).
[7] Kim, S. B., Ozawa, T., Watanabe, S., Umezawa, Y. High- throughput sensing and noninvasive imaging of protein nuclear transport by using reconstitution of split renilla luciferase. PNAS, 101, 11542–11547 (2004).