"
"
Team:Oxford/biosensor characterisation
From 2014.igem.org
(Difference between revisions)
Olivervince (Talk | contribs) |
(proofread) |
||
| Line 481: | Line 481: | ||
<img src="https://static.igem.org/mediawiki/2014/4/46/Oxford_data2.png" style="float:right;position:relative; width:80%;margin-left:10%;margin-right:10%;" /> | <img src="https://static.igem.org/mediawiki/2014/4/46/Oxford_data2.png" style="float:right;position:relative; width:80%;margin-left:10%;margin-right:10%;" /> | ||
<br><br> | <br><br> | ||
| - | We then ran the model for the correct amount of time (2 hours 20mins incubation with ATC) and ran it for lots of different concentrations of ATC over the range that the wet- | + | We then ran the model for the correct amount of time (2 hours 20mins incubation with ATC) and ran it for lots of different concentrations of ATC over the range that the wet-lab team tried. The parameters are still arbitrary at this point (the same as above) and the results of the graphs are therefore arbitrary are as well, but the input values are now correct. The graphic below shows how we used the existing model to obtain the same graphs as the wet-lab team had obtained. |
<br><br> | <br><br> | ||
The numerical inputs that were used to model this data set were therefore: | The numerical inputs that were used to model this data set were therefore: | ||
| Line 494: | Line 494: | ||
<h1>Parameters</h1> | <h1>Parameters</h1> | ||
| - | As all results are arbitrary up to this point, it is now time to calculate the parameters that will make the model’s response match up with the wet-lab data. The purpose of doing this is | + | As all results are arbitrary up to this point, it is now time to calculate the parameters that will make the model’s response match up with the wet-lab data. The purpose of doing this is that the model will be able to give relatively accurate predictions of the response of the bacteria to further testing, therefore making the development of the biosensor much more efficient. The amount of data here will not allow us to calculate the parameters to a high level of accuracy, but it should be able to give us some very good approximations of what we can expect. |
<br><br> | <br><br> | ||
| Line 507: | Line 507: | ||
<li>β1 = Basal transcription rate of <font style="font-style: italic;">dcmR</font></li> | <li>β1 = Basal transcription rate of <font style="font-style: italic;">dcmR</font></li> | ||
| - | Remember that because the mCherry gene is tagged (translational fusion) onto the end of the <font style="font-style: italic;">dcmR</font> gene, <font style="font-style: oblique;"> the mCherry fluorescence will be the same as the amount of DcmR protein present</font>. However, there is not very comprehensive data in the literature about the values that we can expect from the behaviour of the <font style="font-style: italic;">dcmR</font> gene. | + | Remember that because the mCherry gene is tagged (translational fusion) onto the end of the <font style="font-style: italic;">dcmR</font> gene, <font style="font-style: oblique;"> the mCherry fluorescence will be the same as the amount of DcmR protein present</font>. However, there is not very comprehensive data in the literature about the values that we can expect from the behaviour of the <font style="font-style: italic;">dcmR</font> gene and its stability in vivo. |
<br><br> | <br><br> | ||
Revision as of 10:58, 14 October 2014









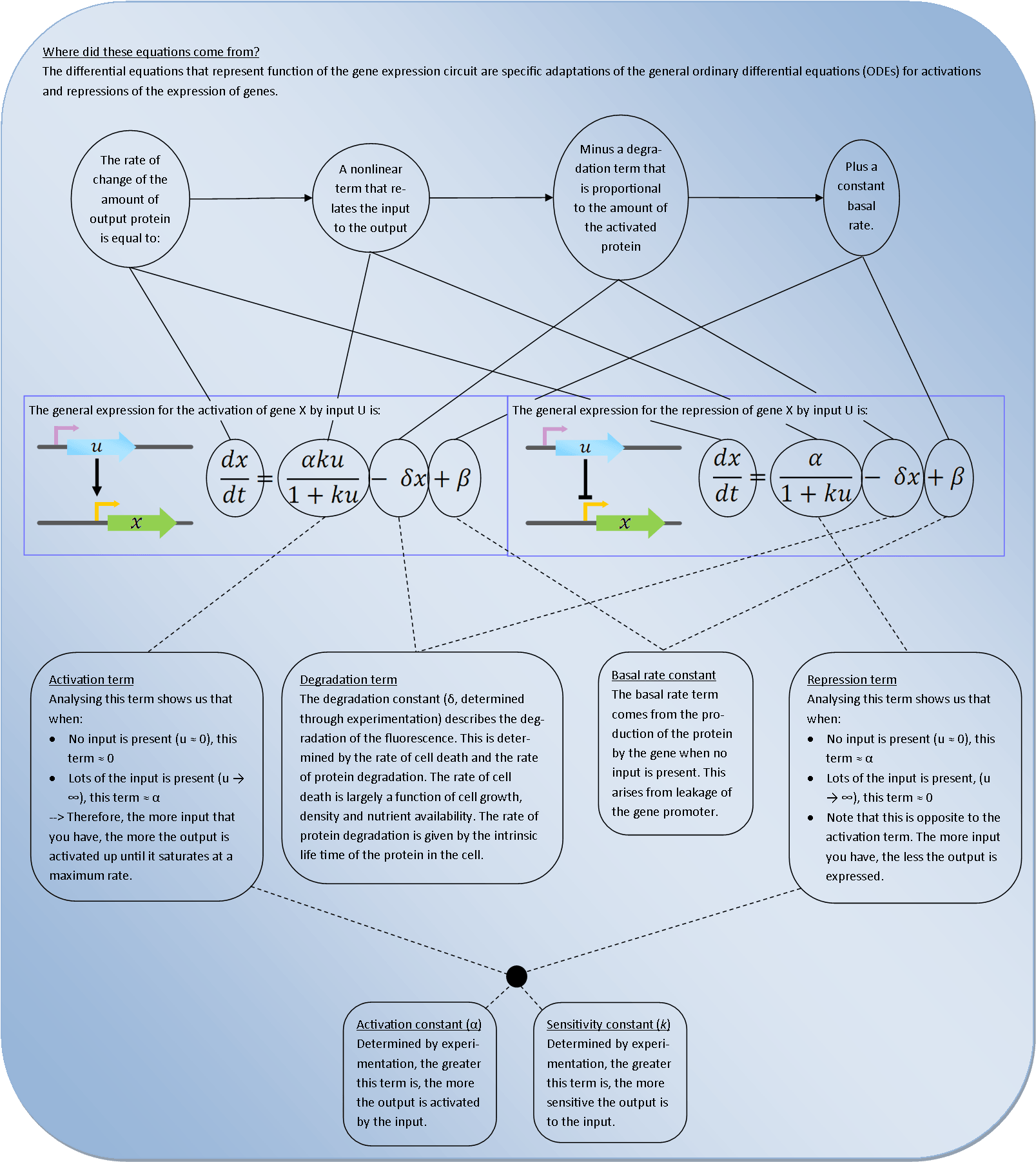
 Deterministic models are very powerful tools for synthetic biology. They describe the behaviour of the bacteria at the population level and use Ordinary Differential Equations (ODEs) to relate each activation and repression. By constructing a cascade of differential equations one can build a realistic model of the average behaviour of the system.
Deterministic models are very powerful tools for synthetic biology. They describe the behaviour of the bacteria at the population level and use Ordinary Differential Equations (ODEs) to relate each activation and repression. By constructing a cascade of differential equations one can build a realistic model of the average behaviour of the system.


 Oxford iGEM 2014
Oxford iGEM 2014