 "
"
Team:Marburg:Project:NRPS
From 2014.igem.org
RiboSURF

The aim of our project was to make it possible to combine the advantages of the nonribosomal peptide synthesis with the ribosomal pathway, i.e. the enormous repertoire of amino acids combined with the ability of the ribosome to synthesize huge proteins.
Introduction
The ribosome is the primary site of protein synthesis in a cell with its advantages being the high reliability of protein synthesis combined with the possibility of assembling huge proteins from amino acids using mRNA as blueprint. As an adaptor that guides the correct incorporation of amino acids as specified in the mRNA template ribosomes use aminoacyl-tRNAs. These are generated by activation of the needed amino acid using ATP. The created adenylate intermediate is then transferred to the 3’ terminus of the corresponding tRNA. This loading reaction of tRNA is catalysed by an enzyme family of aminoacyl-tRNA synthetases (aaRSs) (Rodnina, 2013). These form multi-synthetase complexes (MSCs) in eukaryotes and archaea that contain not only the aaRSs themselves but also accessory protein factors for the local scaffolding of tRNAs to the aaRS (Quevillon et al., 1997). One of these accessory proteins, Arc1p (Figure 2), has been shown to improve the functionality of the MSC in Saccharomyces cerevisiae by facilitating tRNA binding (Simos et al., 1998).
By contrast to the ribosome, the nonribosomal peptide synthesis occuring in the secondary metabolism of many microbes on the other hand uses modular multi enzyme complexes called nonribosomal peptide synthetases (NRPSs). Due to the fusion of different domains nature build highly developed catalysts for the production of a huge amount of biologically active compounds in evolutionary time (Strieker et al., 2010). The reactions that these modules catalyse can be compared to equivalent steps in the ribosomal pathway. Especially interesting in this context is the first step catalysed by an adenylation (A) domain which is the activation of the corresponding amino acid using ATP to yield an adenylate intermediate and thus closely resembles the corresponding step of the ribosomal pathway (Sieber and Marahiel, 2005). However, in contrast to the limited repertoire of amino acids the ribosome uses there are A-domains known that can select from hundreds of proteinogenic and non-proteinogenic building blocks. Nevertheless the peptides that are assembled seem to be limited in their size to about 50 amino acids (Caboche et al., 2008). Intrigued by the multitude of possibilities these A-domains offer we wanted to make their utilization within the ribosomal pathway possible to overcome the size limit of NRPSs.
To reach that aim, we planned to create a fusion protein that has the capability to activate amino acids derived from NRPSs’ A-domains and tRNA binding capabilities derived from aaRSs.
 Figure 1: Model of the RiboSURF.
Figure 1: Model of the RiboSURF.
Structural characterization of the Arc1p-C tRNA binding domain
Our choice for these two domains were on the one hand PheA (BBa_K1329005) from gramicidinS synthetase which was already well characterized and has been shown to activate L- and D-phenylalanine also in a non-native context (Stevens et al., 2006) which is a necessary criterion for the construction of our enzyme. On the other hand the search for a domain that conferred tRNA binding capabilities faced the challenge that the tRNA binding domain is often established by the correct spatial arrangement of parts of the peptide chain. Finally; in the case of the C-terminal part of Arc1p (Arc1p-C) (BBa_K1329004) from S. cerevisiae, we found a domain that shows these binding capabilities in one peptide strand and thus was accessible for further engineering by the introduction to our fusion protein. However, structural information on this domain and a structural understanding of how this domain interacts with tRNA was crucially missing. Therefore, we crystallized Arc1p-C (Figure 2A) and determined its crystal structure to 1.8 Å resolution (Figure 2B). Data were collected at ID23-1 at the European Synchrotron Radiatian Facility, Grenoble, France.
 Figure 2: Structure determination of Arc1p-C. (A) Crystals of
Arc1p-C were obtained after 2 to 4 days in a wide range of crystallization conditions.
(B) Crystal structure of Arc1p shown as ribbon (left) – and electrostatic surface representation (right). Dashed lines indicate the putative tRNA binding site.
This fact is underlined by the presence of a glycerol from the cryosolution because glycerol likely mimics ribose sugar of a RNA.
Figure 2: Structure determination of Arc1p-C. (A) Crystals of
Arc1p-C were obtained after 2 to 4 days in a wide range of crystallization conditions.
(B) Crystal structure of Arc1p shown as ribbon (left) – and electrostatic surface representation (right). Dashed lines indicate the putative tRNA binding site.
This fact is underlined by the presence of a glycerol from the cryosolution because glycerol likely mimics ribose sugar of a RNA.
Molecular modelling the Arc1p interaction with tRNA
To understand the binding mode of tRNA to Arc1p-C we used the DUCK-BP server employing the FTdock algorithm (Gabb et al., 1997) to calculate possible binding modes (Figure 2). Taking functional aspects into account only the highest scoring docking model seems to represent the real binding mode since all the others lead to a blockade of the tRNA acceptor stem. Furthermore, solution 1 perfectly matches with the presence of a glycerol potentially mimicking a RNA backbone ribose position (see also Figure 2B).
 Figure 3: Calculated docking modes of tRNA to Arc1p-C.
Only the first one seems sensible from a functional point of view because all other solutions
hide the 3’ end of the tRNA to which canonical aaRSs transfer amino acids.
Figure 3: Calculated docking modes of tRNA to Arc1p-C.
Only the first one seems sensible from a functional point of view because all other solutions
hide the 3’ end of the tRNA to which canonical aaRSs transfer amino acids.
Structure-based design of Arc1p-PheA catalysts
In order to create that correct distance between those two domains we wanted to use computational methods to simulate the protein with different linker lengths between the two domains. As a linker we used repeated GSSG units. With the crystal structure of PheA known we still needed to solve the crystal structure of Arc1p-C and find the binding mode of the tRNA to Arc1p-C in order to build a precise molecular model of rationally design suitable linker lengths that balance between on the one hand enough flexibility for favourable positioning of tRNA and activated amino acid and a sufficient increase in local reactant concentration for efficient catalysis to occur on the other hand. The results of the simulation of 0, 1, 2, 4 and 8 GSSG units suggested that the best distance that should lead to the highest catalyst activity is reached with two GSSG units (Figure 4). For the testing of our prediction, we synthesized the fusion protein with these five different linker lengths.
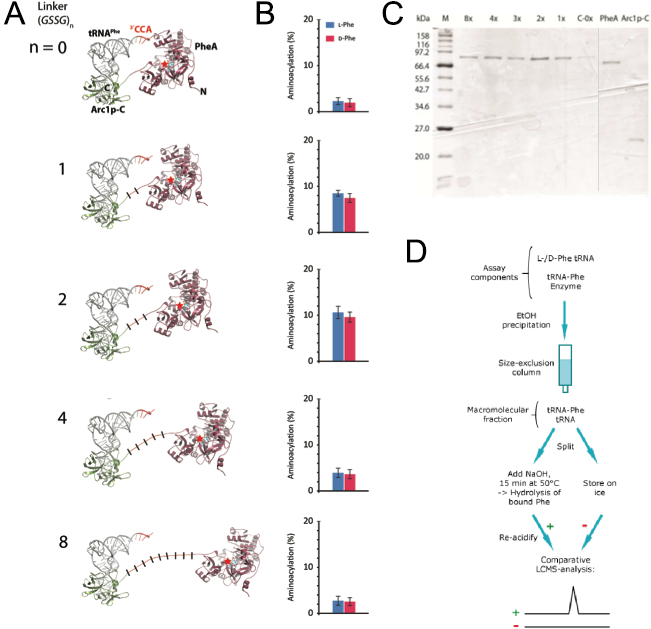 Figure 4: Design and functional analysis of different RiboSURF constructs. (A) Computational molecular model of Arc1p-C-PheA fusion proteins with
different linker lengths, and (B) their activities in aminoacylating tRNA. (C) Coomassie-stained SDS-PAGE of purified RiboSURF catalysts proteins employed in this project.
Proteinmarker: Broad Range (New England Biolabs). (D) Workflow for the detection of aminoacylation levels.
Figure 4: Design and functional analysis of different RiboSURF constructs. (A) Computational molecular model of Arc1p-C-PheA fusion proteins with
different linker lengths, and (B) their activities in aminoacylating tRNA. (C) Coomassie-stained SDS-PAGE of purified RiboSURF catalysts proteins employed in this project.
Proteinmarker: Broad Range (New England Biolabs). (D) Workflow for the detection of aminoacylation levels.
The 8x linker was constructed from a synthesized template and integrated into pET28a by Gibson assembly. The other lengths were created by round-the-horn site directed mutagenesis using phosphorylated primers and a ligation after the PCR. All the proteins (Figure 4C) were produced as His6 tagged fusion proteins. For testing the catalytic activity of each mutant we used an LC-MS based assay. Since the phosphate backbone leads to a multiple negative charge the direct analysis by mass spectrometry probably would lead to a too poor sensitivity to discriminate loaded and unloaded tRNAs (Figure 4D). Thus we decided to make an indirect measurement based on an established method. After the reaction mixture was incubated we separated the tRNA from all the other components by size-exclusion chromatography. The obtained mixture of loaded and unloaded tRNAs was split and one half kept on ice while the other was treated with a base to cleave the oxoester bond. Afterwards the amount of released amino acid in the base treated sample was detected by LC-MS using the untreated sample as a negative control.
Measurements showed that all fusion constructs were able to load L- as well as D-phenylalanine onto tRNAPhe. As the computational molecular model already suggested the varying linker length showed a clear influence on the yield levels with the 2x-GSSG linker showing the highest catalytic activity yielding 11% loaded tRNA after 30 min while the 8x-construct reached only a maximum of 3% loaded tRNA as well as the linker-less version. The remaining constructs showed intermediate results in good correspondence with the computational model (Figure 4B).
 Figure 5: Negative and positive controls and measured timecourse of the aminoacylation.
Figure 5: Negative and positive controls and measured timecourse of the aminoacylation.
Furthermore the linking of the two domains leading to the increase in reactant concentration and the correct spatial arrangement is indeed important for the catalytic effect to occur since a mixture of the unlinked domains showed only background levels of aminoacylation (Figure 5). Further negative controls included testing the reaction without enzyme or ATP. As a positive control to evaluate the method phenylalanyl-tRNA synthetase (PheRS) was used. A time-dependent measurement of the aminoacylation level showed that a maximum is reached after 30 min. To test if other tRNAs except for the tRNAPhe can be aminoacylated using our fusion construct we carried out aminoacylation assays with five additional Escherichia coli tRNAs (Figure 6). The measurements suggest in agreement with previous studies that all tRNAs were loaded similarly well.
 Figure 6: Aminoacylation activity of the 2x linker construct using different tRNAs relative to the activity observed using tRNAPhe and L-Phe.
Figure 6: Aminoacylation activity of the 2x linker construct using different tRNAs relative to the activity observed using tRNAPhe and L-Phe.
Improve a brick
Nonribosomal peptide synthetases (NRPSs) are a most fascinating subject because of the huge amount of biologically active compounds they produce that show a broad spectrum of clinical applications that even includes their use as last resort antibiotics, antitumor or antifungal agents and immunosuppressants. These interesting properties especially derive from the nonribosomal peptide synthetases unique feature to incorporate non-proteinogenic features such as heterocycles, fatty acids, macrocycles, D-amino acids and other non-proteinogenic amino acids. Last years’ team of the university Heidelberg used nonribosomal peptide synthetases of Brevibacillus parabrevis in order to synthesize a tripeptide by interchanging modules of an already existing NRPS cluster. The problem of previous applications such as these is that the size of synthesized peptides is limited. Even naturally occurring peptides synthesized nonribosomally seem to have an upper limit of 50 amino acids (Caboche et al., 2008). Therefore it would be great to establish a system that uses these capabilities to introduce non-proteinogenic amino acids but is able to overcome the size limit of the synthesized peptides. That is why we improved the brick BBa_K1152005 which contains the expression cassette for NRPS synthesizing a Phe-Orn-Leu-tripeptide. In RiboSURF, we improved the A-domain that activates phenylalanine (BBa_K1329005) in that nonribosomal peptide synthetase. Our improvement makes it possible to use the NRPS module even in its non-natural environment isolated from its NRPS context. By the fusion of the module from NRPS to a tRNA binding domain the catalytic activity of this NRPS module was introduced to the ribosomal pathway which has much more powerful capabilities regarding the protein synthesis as it can assemble huge proteins using RNA as a blueprint.
Caboche, S., Pupin, M., Leclère, V., Fontaine, A., Jacques, P., and Kucherov, G. (2008) NORINE: a database of nonribosomal peptides. Nucleic Acids Res 36: D326–31 http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2238963&tool=pmcentrez&rendertype=abstract. Accessed October 14, 2014.
Gabb, H.A., Jackson, R.M., and Sternberg, M.J. (1997) Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol 272: 106–20 http://www.ncbi.nlm.nih.gov/pubmed/9299341. Accessed October 14, 2014.
Quevillon, S., Agou, F., Robinson, J.-C., and Mirande, M. (1997) The p43 Component of the Mammalian Multi-synthetase Complex Is Likely To Be the Precursor of the Endothelial Monocyte-activating Polypeptide II Cytokine. J Biol Chem 272: 32573–32579 http://www.jbc.org/cgi/doi/10.1074/jbc.272.51.32573. Accessed October 14, 2014.
Rodnina, M. V (2013) The ribosome as a versatile catalyst: reactions at the peptidyl transferase center. Curr Opin Struct Biol 23: 595–602 http://www.ncbi.nlm.nih.gov/pubmed/23711800. Accessed October 14, 2014.
Sieber, S.A., and Marahiel, M.A. (2005) Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev 105: 715–38 http://www.ncbi.nlm.nih.gov/pubmed/15700962. Accessed October 14, 2014.
Simos, G., Sauer, A., Fasiolo, F., and Hurt, E.C. (1998) A conserved domain within Arc1p delivers tRNA to aminoacyl-tRNA synthetases. Mol Cell 1: 235–42 http://www.ncbi.nlm.nih.gov/pubmed/9659920. Accessed October 14, 2014.
Stevens, B.W., Lilien, R.H., Georgiev, I., Donald, B.R., and Anderson, A.C. (2006) Redesigning the PheA domain of gramicidin synthetase leads to a new understanding of the enzyme’s mechanism and selectivity. Biochemistry 45: 15495–504 http://www.ncbi.nlm.nih.gov/pubmed/17176071. Accessed October 14, 2014.
Strieker, M., Tanović, A., and Marahiel, M.A. (2010) Nonribosomal peptide synthetases: structures and dynamics. Curr Opin Struct Biol 20: 234–40 http://www.ncbi.nlm.nih.gov/pubmed/20153164. Accessed July 22, 2014.