 "
"
Team:Freiburg/Content/Notebook/Labjournal
From 2014.igem.org
Cloning
Cloning - May
Cloning - June
Cloning - July
Cloning - August
Cloning - September
Cloning - October
Viral Vectors
Viral Vectors - May
2014/05/21
Transfection/ Virus production
For virus production Phoenix cells (producer cell line) were splitted (well separated) on 100mm plates. At 70% cell density cells were transfected using polyethylenimine.
- remove medium and refill with 5 ml new completed growth medium (DMEM)
- 600 µl transfection mastermix was prepared (8 µg pMIG IRES EGFP, 24 µl PEI, rest OptiMEM)
- mastermix was incubated 15 min and carefully drop on the plates
Plates were incubated at 37°C. The supernatant after 24 was removed and refilled with 5 ml new DMEM, the supernatant was collected after 48 h (refilled with 5 ml DMEM) as well as 72 h.

Phoenix cells transfected with pMIG IRES EGFP one day after transfection.
2014/05/25
Transduction mouse cells
NIH 3T3 cells (60% density) were transduced with MuLV IRES EGFP.
- 500 µl of supernatant was removed
- 1 µl Polybrene was added (10mg/ml)
- 500 µl virus supernatant was added (sterile filtered)
- incubation at 37°C for 6h
- cell supernatant was replaced with fresh DMEM
- transduction was repeated once
Pictures could be made after 48 h of incubation.

NIH 3T3 cells were transduced twice with MuLV IRES EGFP for 6h. Picture was made after 48 h of incubation at 37°C.
Viral Vectors - June
2014/06/20
Thawing of eukaryotic cells
New Phoenix cell stocks were thawed:
- cryotube was thawed at 37°C water bath until almost defrosted
- stock was filled in 9 ml warm completed growth medium and centrifuged at 900 rpm for 2 min
- medium was removed and refilled with 10 ml warm completed growth medium
- cells were seeded on 100 mm plates
Testing optimal cell density of mouse fibroblasts
NIH 3T3 have a really fast growth so that we tested the optimal cell number for seeding NIH 3T3 for having around 60% cell density on the next day. NIH 3T3 cells grow very fast; therefore we have tested the optimal seeding cell number to obtain 60% cell density on the next day. Results indicate that the optimal cell number is 1 &ndash 1.5x10^5 cells per well ( = 0.5 – 0.75 cells/ml)

2014/06/22
Transfection/ Virus production
Transfection of Phoenix cells (70% density) with pMIG IRES EGFP (protocol: 2014/05/21) (2 x 100mm plate)

Phoenix cells transfected with pMIG IRES EGFP.
2014/06/24
Transfection/ Virus production
Transfection of Phoenix cells (70% density) with pMIG IRES EGFP (protocol: 2014/05/21) (5 x 100mm plate)
2014/06/27
Thawing new HEK 293 cells
(protocol: 2014/06/20)
Transfection CHO cells with receptor
Transfection of CHO cells with SLC7a1 (for later transduction with virus). Medium was changed after 5 h. Cells were incubated for 24 h before viral transduction with MuLV IRES EGFP, medium change after 16 h.


(left) Cho cells without receptor were transduced with MuLV IRES EGFP ,(right) CHO cells transfected with SLC7a1 and transduced with MuLV IRES EGFP (24h after transfection). Analyses with flow cytometry indicates that 5% of cells were transfected (transfection control) and 2% of cells transfected with the receptor were infected with MuLV.

2014/06/27
Transduction mouse cells (different incubation times)
NIH 3T3 cells (60% density) were transduced with MuLV IRES EGFP and incubated for 8, 16, 24 and 2 x 8 hours. Virus was taken from different supernatants (an older one and a newer one) to see, if it makes any difference. Cells were infected with supernatant (500µl viral supernatant, 500µl completed growth medium + 1µl Polybrene/ml) harvested at different time points. Results indicate that there was no difference between older and newer virus; best results were given with an infection time of 2 x 8 hours.

For testing, if centrifugation brings better transduction efficiencies, mouse cells were infected with the different viral supernatants and centrifuged for 45 min, 1800 rpm, 32°C. In two wells it was tested if the double amount of Polybrene brings better transduction efficiencies. However, we found out that cells were death after centrifugation.

2014/06/30
Transfection CHO cells with receptor
CHO cells were transfected with the receptor (for later transduction). Medium was removed and filled with 2 ml new medium per well. Medium was changed after 5 h. Cells were transduced with MuLV IRES EGFP after 24 h of incubation at 37°C.

CHO cells were transduced with MuLV IRES EGFP 24 h after transfection with SLC7A1.
Transfection/ Virusproduction
Phoenix cells were transfected with pMIG IRES EGFP (protocol: 2014/05/21).
Viral Vectors - July
2014/07/03
Transfection CHO cells with receptor
CHO cells were transfected with the receptor (for later transduction). Medium was removed and filled with 2 ml new medium per well. Medium was changed after 5 h. Cells were transduced with MuLV IRES EGFP after 24 h of incubation at 37°C. This time there were no results du to high density of cells during transduction.
Freezing (cryopreservation) of eukaryotic cells
Phoenix cells were frozen at -80°C.
- removal of medium and washing with cold PBS
- addition of 1 ml 0,05% Trypsin per plate, incubation for 1-2 min)
- stopping of reaction with 5 ml DMEM (with FCS)
- centrifugation (5 min, 900 rpm)
- removal of supernatant and resuspension in 2 ml FCS (+10% DMSO)
- quick transfer in steril cryotube (1ml per tube) and quick freezing in -80°C
2014/07/06
Transfection CHO cells with receptor
CHO cells were seeded on cover slips and transfected with the receptor (for later transduction). Due to the fact that cells must be in growth phase during transduction with virus the cell density was set to 40%. Medium was changed after 5 h. Cells were transduced with MuLV IRES EGFP after 24 h of incubation at 37°C.
CHO cells transfected with SLC7a1 and transduction with MuLV IRES EGFP 24 h after transfection (A) experimental scheme. CHO cells were transduced with MuLV IRES EGFP 24 h after transfection with SLC7a1. (B) Transduced CHO cells expressing EGFP.
2014/07/08
Phoenix cells were transfected with pMIG IRES EGFP (protocol: 2014/05/21)

FACS results of pMIG IRES EGFP transfected Phoenix cells. Phoenix cells were transfected with pMIG IRES EGFP for production of viral particles. They were anaylsed by flow cytometry after three days of virus production (middle and upper pictures); negative control (upper picture).
2014/07/10
Transfection of CHO cells with receptor
CHO cells were transfected with the receptor (for later transduction). Medium was changed after 5 h and cells were transduced with MuLV IRES EGFP. This experiment gave no results.

2014/07/10
Fixation of cells on cover slips
CHO cells (transfected with SLC7a1; 2014/07/04) were fixed on cover slips
- Medium was removed and cells were washed with PBS
- Appropriate amount of 4% PFA/PBS was added (200µl on 24W) and incubated for 10 min on ice
- PFA was removed and plate was washed with PBS
- Cover slips were fixed with Mowiol on slides
2014/07/11
Transfection of HEK cells with receptor
HEK 293 cells were transfected with the receptor (for later transduction). Medium was changed after 5 h. Cells were transduced with MuLV IRES EGFP after 24 h of incubation at 37°C.
HEK293 transfected with SLC7a1 and transduction with MuLV IRES EGFP 24 h after transfection.
2014/07/14
Transfection/ Virus production

FACS results of pMIG IRES EGFP transfected Phoenix cells. Phoenix cells were transfected with pMIG IRES EGFP for production of viral particles. They were anaylsed by flow cytometry after three days of virus production (middle and upper pictures); negative control (upper picture).
2014/07/17
Transduction of mouse cells
Different volumes of virus supernatant were added to mouse cells (on 24W plate, 70% density) and analyzed by FACS (green), Microscopy (yellow) and Western Blot (blue) after 48 h.

green: anaylsis with flow cytometry
NIH cells transduced with MuLV IRES EGFP (left: 0,5 ml Virus + 0,5 ml DMEM; middle: 0,75 ml Virus + 0,25 ml DMEM; right: 1 ml Virus + 0 ml DMEM). Pictures made after 48 h.
yellow: fixation with PFA on cover slips
- Removal of medium
- Washing with cold PBS
- Adding of 400 µl PFA and incubation for 10 min on ice, another 10 min at RT
- Incubation of cover slips for 10 sec in DAPI solution (1:5000 in water)
- Washing in water
- Mounting with Mowiol on slides
Cells detach from the cover slip, therefore a coating is necessary e.g. with Poly-L-Lysine no results (better use poly-lysine for better grip of cells on cover slip)
blue: preparation for Western Blot via RIPA Lysis (as positive control for anti-CAT1 antibody)
- Removal of medium
- Washing with ice cold PBS
- Addition of 100 µl RIPA Buffer (completed with Phosphatase-Inhibitor-Mix)
- Incubation 10 min on ice
- Removal of cells with tip and transfer into Eppendorf tube
- Incubation for 10 min on ice
- Centrifugation for 5 min 10000 x g
- Transfer of 60 µl supernatant in new tube
- Addition of 15 µl 5 x SDS loading dye (with β-Mercaptoethanol)
- Cooking for 10 min at 95°C or for 15 min at 72°C
- Freezing at -24°C
Transfection of CHO cells with receptor
CHO cells growing in completed HTS medium (K1) were compared to CHO cells growing in completed DMEM medium. Cells were transfected with the receptor. Afterwards both kinds of CHO cells were infected with MuLV IRES EGFP and analyzyd using flow cytometry investigate which cells are better for transfection and transduction. Medium was changed after 5 h. Cells were incubated for 24 h at 37°C.




FACS results of CHO HTS (left) and CHO DMEM (right) cells transfected the receptor and transduced with MuLV EGFP.
2014/07/21
QR-code on 96W plate
HEK cells were transfected with the blue light system with SEAP as reporter and seeded on a 96W plate for pattern generation.
- Transfection of HEK293 in suspension (2 x 10^5 cells/ml, 100µl transfection mix/ml) with PKM292, PKM297 and PKM084 (1,5 µg DNA/ml cell suspension)
- Incubation for 5 h at 37°C and seeding on a 96W plate (with photo mask) afterwards (120µl/well)
- After 24 h of incubation at 37°C cells were illuminated with blue light for 5 h.
- Again after 24 h of incubation a SEAP assay was made directly in the plate:
- 30 min incubation of plate at 65°C
- addition of 100µl SEAP buffer into each well (ca. 90µl medium was left in each well)
- before measurement 20 µl substrate was added

HEK293 cells were transfected with the blue light system (PKM292, PKM297) and light induced SEAP as reporter (PKM084). Single wells were covered with a photo mask to prevent cell from light exposure. 24 hours after illumination with blue light a SEAP assay was performed leading to colour changing of wells containing SEAP. A photo mask in heart form was used. (B) a pink colour was set to green and a yellow colour was set to blue at a computer, (C) the same experiment was performed with multiple well plates with separated wells, (D) the contrast an the colours of the wells were changed at a computer. All wells together are forming the word: IGEM.

A pink colour was set to green and a yellow colour was set to blue at a computer.
Transfection/ Virus production
Phoenix cells were transfected with pMIG IRES EGFP and pMIG EGFP(protocol: 2014/05/21)

FACS results of pMIG IRES EGFP transfected Phoenix cells. Phoenix cells were transfected with pMIG IRES EGFP for production of viral particles. They were anaylsed by flow cytometry after three days of virus production (middle and upper pictures); negative control (upper picture).
2014/07/31
Improvement of Transduction
Transduction of NIH 3T3 cells with two different viral supernatants via three different methods.
- 1.2 µl Polybrene adding directly to 1 ml DMEM on cells and adding 1 ml viral supernatant afterwards
- 2.addition of 1 µl Polybrene to 1 ml viral supernatant and addition of the mixture to 1 ml DMEM on the cells
- 3.addition of 2 µl Polybrene to 1 ml viral supernatant and addition of the mixture to 1 ml DMEM on the cells
- Incubation for 48 h at 37°C

NIH3T3 cells were transduced with MuLV EGFP (upper pictures) and with MuLV IRES EGFP (lower pictures) left: transduction method 1; middle: transduction method 2; right: transduction method 3
Transduction of NIH3T3 cells with two different viral supernatants (MuLV IRES EGFP or MuLV EGFP) with either 1 or 2 µl Polybrene.
- 1. 1 µl Polybrene added directly to 1 ml DMEM on cells, addition of 1 ml virus supernatant
- 2. 2 µl Polybrene added directly to 1 ml DMEM on cells, addition of 1 ml virus supernatant
- 3. 2µl Polybrene was added to 1 ml viral supernatant; mixture was added to 1 ml DMEM
Centrifugation at 37°C for 45 min at 400 RPM. Results indicate that cells do not like to be centrifuged.


NIH3T3 cells transduced with MuLV EGFP (A + B) and MuLV IRES EGFP (C + D) either with 1 µl (A + C) or 2 µl (B + D) Polybrene and centrifuged 45 min at 37°C for 400 RPM
Testing difference in transfection efficiency of Phoenix and HEK cells/ negative control for MuLV
Phoenix cells and HEK cells were tested for there transfection capacity and compared. In addition, it was tested that the virus cannot infect Phoenix nor HEK cells. So both kinds of cells were transfected with PHB308 (mCherry, 3 µg/well) and in parallel transduced with MuLV IRES EGFP (1 ml/well + 2 µl Polybrene).


HEK cells (A + B): transduced with MuLV IRES EGFP (A); negative control (B); Phoenix cells (C + D): transduced with MuLV IRES EGFP (C); negative control (D); HEK cells (E) and (F) Phoenix cells transfected with PHB308 and analysed after 24 h.
Comparison of transfection capacity of HEK 293 cells (red) and Phoenix cells (blue). Both cell lines were transfected with 3 µg PHB308 (mCherry) per well (6W plate) and analysed via FACS after 24 h.
Viral Vectors - August
2014/08/01
Generation of a GFP mouse cell line
For testing whether MuLV can stable transfer genes into cells, a stable mouse cell line using this virus was generated.
Therefore two 100mm plates were transduced with 3 ml virus supernatant (MuLV IRES EGFP) and splitted as usual. Cells were sorted with a cell sorter. Analysis via FACS happened before and after sorting. The analysis was repeated after several rounds of splitting.

NIH3T3 cells transduced with MuLV IRES EGFP and sorted. Negative control (left), before sorting (middle), after sorting (right).
2014/08/03
Testing different transfection methods with different cells
For optimizing transfection in different cell lines transfection methods and different concentrations of the transfection mixtures were compared. The experiment was done with mouse cells (NIH3T3), hamster cells (CHO) and human cells (Phoenix). As transfection reagents lipofectamin and PEI were used in different concentrations.

NIH3T3 cells transduced with MuLV IRES EGFP and sorted. Negative control (left), before sorting (middle), after sorting (right).
Transfection with Lipofectamin (for 3 wells):
- (solutions A) 50 µl OptMEM was mixed with either:
- 1. 1 µl Lipofectamin + 1,5 µl PHB308
- 2. 2,5 µl Lipofectamin + 1,5 µl PHB308 or
- 3. 4 µl Lipofectamin + 1,5 µl PHB308
- incubation for 25 min at RT
- (solution B) 150 µl OptiMEM was mixed with 1,5 µl PHB308 (2,5 µg/µl) and 4 µl Plut Reagent
- incubation for 15 min
- solutions A (1-3) were then mixed with 50 µl of solution B and incubated for 5 min at RT
- 100 µl of transfection solution were added to each well
- no medium changing, incubation at 37°C for 24 h
Transfection with PEI (for 1 well):
- 0,2 µl PHB308 was mixed with 40 µl OptiMEM and DNA, for each well another concentration of DNA was added:
- 1. 1,5 µl PEI
- 2. 3 µl PEI
- 3. 5 µl PEI
- Incubation for (optimal) 10,8 min
- addition of 40 µL solution to each well
- medium changing after 5 h of incubation at 37°C, incubation for 24 h at 37°C

Pictures and FACS data of different transfection methods with different kinds of cells. A: Phoenix cells transfected with PHB308 (mCherry) via 1 µl (left), 2,5 µl (middle) and 4 µl Lipofectamin; B: Phoenix cells transfected with PHB308 (mCherry) via 1,5 µl (left), 3 µl (middle) and 5 µl Pei; C: Cho cells transfected with PHB308 (mCherry) via 1 µl (left), 2,5 µl (middle) and 4 µl Lipofectamin; D: Cho cells transfected with PHB308 (mCherry) via 1,5 µl (left), 3 µl (middle) and 5 µl Pei; E: NIH3T3 cells transfected with PHB308 (mCherry) via 1 µl (left), 2,5 µl (middle) and 4 µl Lipofectamin; F: NIH3T3 cells transfected with PHB308 (mCherry) via 1,5 µl (left), 3 µl (middle) and 5 µl Pei.
2014/08/06
Transfection of HEK cells with receptor
HEK293 were transfected with the receptor plus mCherry to see, if the virus infect only cells expressing the receptor (the two plasmids should infect same cells). In parallel cells were transfected with the blue light and the red light positive control (PMZ422 and PSAM200) and the receptor inducible by the light systems. After 24 h cells (expressing the receptor) were infected with MuLV IRES EGFP.

HEK 293 tranfected with PHB308 (mCherry) and the receptor. Transduction with MuLV after 24 h of incubation. Pictures were taken two days post transfection.
Transfection HEK and Pheonix cells with receptor (transfection while seeding)
To avoid a too high cell density for transduction cells were transfected while seeding. The PEI transfection mix was given to the HEK or Phoenix cell suspension (1,5 x 10^5 c/ml, 100 µl mix per 1 ml cells) and seeded on a 12W plate. Results indicated that PEI was too toxic for cells during this method.

2014/08/09
Virus dilution
Virus supernatant was diluted with fresh DMEM before transduction. Cells were analyzed after 48 h.
- 0,5 ml virus + 0,5 ml DMEM
- 0,4 ml virus + 0,6 ml DMEM
- 0,3 ml virus + 0,7 ml DMEM
- 0,2 ml virus + 0,8 ml DMEM
- 0,1 ml virus + 0, 9 ml DMEM

NIH3t3 cells transduced with MuLV IRES EGFP in different dilutions (upper from left to right: 5/10, 4/10, 3/10, 2/10, 1/10 virus supernatant. Pictures were made after 48 h of incubation at 37°C.

NIH3t3 cells transduced with diluted MuLV IRES EGFP and analysed by FACS after 48 h, red: 1/10, blue: 2/10, orange: 3/10, green: 4/10, dark green: 5/10 dilution.
2014/08/15
Testing different receptor constructs
HEK293 cells were transfectec with p14rz_004 (labeled with HA-tag), p14ls_006 (labeled with HA-tag and mCherry) and the original receptor plasmid pQCXIN (containing SLC7A1).

Figure : HEK293 cells transfected with different receptor constructs and infected with MuLV EGFP afterwards.
The cells were transfected with pQCXIN (original vector containing SLC7A1) (A), p14rz_004 (labeled with HA-tag)(B) and p14rz_006 (labeled with HA-tag and mCherry)(C). Cells were transduced with MuLV after 24 hours. Pictures were taken after 48 hours. Labjournal
Viral Vectors - September
2014/09/01
QR-code on 96W plate
HEK cells were transfected with the blue light system with SEAP as reporter and seeded on a 96W plate for pattern generation.
- Transfection of HEK293 in suspension (2 x 10^5 cells/ml, 100µl transfection mix/ml) with PKM292, PKM297 and PKM084 (1,5 µg DNA/ml cell suspension)
- Incubation for 5 h at 37°C and seeding on a 96W plate (with photo mask) afterwards (120µl/well)
- After 24 h of incubation at 37°C cells were illuminated with blue light for 5 h.
- Again after 24 h of incubation a SEAP assay was made directly in the plate:
- 30 min incubation of plate at 65°C
- addition of 100µl SEAP buffer into each well (ca. 90µl medium was left in each well)
- before measurement 20 µl substrate was added


left: Supernatants of HEK293 cells transfected with PKM292, PKM297 and PKM084 24 h after illumination with blue light. For visualizing of SEAP a SEAP assay was made (2014-09-03 23.55.54), right: same supernatants with with colour switch that was made after taking picture.
2014/09/03
Virus Half life time
800 µl aliquots of virus supernatants (MuLV IRES EGFP) were incubated for different times at 37°C in the incubator. After incubation cells were infected with the supernatants (250 ml supernatant/ 250 ml medium + 0,5 µl Polybrene). After 48 h cells were anaysed by FACS.
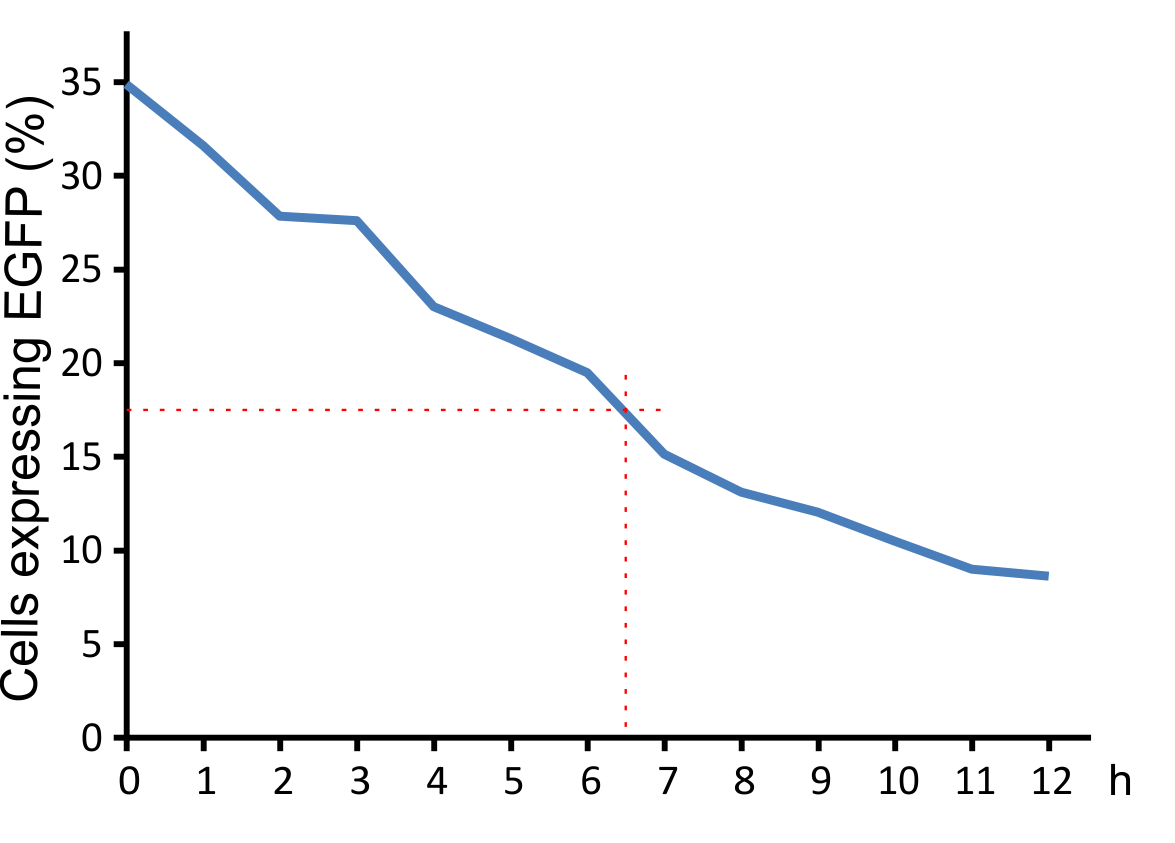
Figure 8: Decay of viral particles.
The virus was first incubated at 37°C and then added to the NIH 3T3 cell culture. Transduction efficiency was measured after 48 hours.
Experiment: receptor life time after splitting
Two 6W of HEK293 cells were transfected with rz006 (receptor labled with mCherry and HA-tag). After each round of splitting cells were analysed by Western blot. Therefore each round one well was splitted 1:2 on two new wells and one well was lysed by Ripa lysis. Afterwards, each lysed sample was frozen in -20°C until use.

Two new cells lines
New cells lines were thawed:
- MCF-7 human breast cancer cells
- A-547 human lung epithel carcinoma
Rezeptorexpression time
HEK293 cells on 16 x 35mm plates were transfected with rz006 (receptor labeled with mCherry and HA-tag). At several time points after transfection cells were lysed with RIPA buffer and analyzed with Western Blot. Samples were frozen at -20°C until analysis.


Western blot of ripa lysed HEK293 cells expressing rz006 (labled with HA-tag). Samples were taken after several time points after transfection.
2014/09/05
Generation of a GFP mouse cell line
For testing whether MuLV can stable transfer genes into cells, a stable mouse cell line using this virus was generated.
Therefore two 100mm plates were transduced with 3 ml virus supernatant (MuLV IRES EGFP) and splitted as usual. Cells were sorted with a cell sorter. Analysis via FACS happened before and after sorting. The analysis was repeated after several rounds of splitting.

Figure 2: Stable integration of EGFP into the genome of the target cells
(A) Histogram of fluorescence intensity at the first sorting step. (B) Histogram of fluorescence intensity at passage xx after sorting. (C) The fraction of fluorescent cells stays constant for many passages.
2014/09/09
Specificity of MuLV
We tested, if MuLV was specific for murine cell lines and was not able to infect cell lines not containing mCAT-1. So we incubated different human cell lines with MuLV EGFP regarding EGFP expression after infection.


Figure 10: Infection of different cell lines with the viral vector derived from the murine leukemia virus.
Cells that were infected by viral particles express EGFP.
2014/09/11
Testing different pMIG constructs
We tested the expression strength of EGFP in pMIG constructs containing only the marker in comparison to constructs containing the target gene upstream of an IRES and EGFP as a marker.


Figure 4: Expression strength of EGFP sorting marker with and without an IRES.
(A) Cargo contains only EGFP, (B) cargo contains EGFP downstream of an IRES, (C) cargo contains a gane of interest, an IRES, and the EGFP marker.
2014/09/26
Transfection/ Virus production (stained virus)
Phoenix cells were stained with DiD, a membrane staining dye.
- Resuspension of 2 µl dye (5mM) in 5 ml OptiMEM
- Removing of medium from five 100mm Pheonix plates and addition of 1 ml staining solution per well
- Incubation for 15 min at 37°C
- Washing of plates two times with DMEM (incubation time between each washing step was at least 10 min
- Transfection of cells with pMIG-mKO, pMIG-BFP2, pMIG-EGFP and pMIG-mKate
- Medium changing after 24 h of incubation at 37°C and refill with 4 ml new DMEM
- Harvest after 48 h and 72 h post transfection
2014/09/28
Virusintegration time in mouse cells
NIH3t3 cells (mouse fibroblasts) were infected with MuLV IRES EGFP at different time points. Directly after the latest infection the cells were splitted on new plates. After 48 h cells were analyzed via FACS.


Figure 3: Integration time of our viral vector into murine cells.
Murine cells were infected with MuLV EGFP and washed at distinct time points.
QR-code on 24W plate (with virus)
For testing the functionality of our system and the functionality of the virus with SEAP, cells were transfected with the blue light system plus light inducible receptor (labled with mCherry) and later infected with MuLV SEAP or MuLV CMV SEAP. Both viruses were compared. In addition, cells were infected with MuLV EGFP as control. Several negative controls were added as well as unilluminated cells expressing the light system for testing leaky expression. Cells were incubated in dark.
- Transfection of cells with eather the blue light system plus receptor (PKM292, PKM297, ls003) or the receptor (rz006 p2a mCherry).
- Cells were transfected via PEI in suspension (3 x 10^5 cells/ml, 100 µl transfection mix/ml, 1,5 µg DNA/ml). Cells were incubated for 5 h at 37°C before centrifugation (900 RPM, 3 min, 24°C). Cells were resuspended with fresh DMEM to a final concentration of 1,5 x 10^5 cells/ml) and seeded (0,5 ml/well).
- Cells were incubated for 24 h at 37°C and the illuminated with blue light for 1 h (not the dark control).
- Cells were infected with 250 µl virus supernatant (+0,5 µl Polybrene) per well. Medium was changed after 4 h of incubation at 37°C.
- After 24 h and after 48 h of incubation 200 µl supernatant was taken and used for SEAP assay as described before.

Since, SEAP is already produced by Phoenix cells during generation of viral particles, SEAP was detected in every negative control.

Blue light induced receptor.
HEK cells were transfected with the blue light system (PKM292 and PKM297) and the light induced receptor (p14ls_003). The receptor was labeled with mCherry. Cells were infected with MuLV EGFP afterwards; (left) incubation in the dark, (middle) after illumination with blue light, (right) cells expressing the light induced receptor were infected with MuLV.
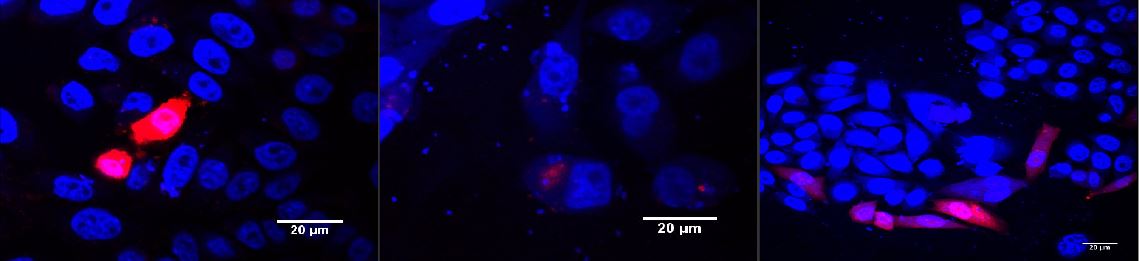
Transduction of mouse cells on cover slips, different colours
Mouse cells (on 25 cm coverslips, covered with polylysine) were infected with MuLV-mKO, MuLV-EGFP, MuLV-BFP2, MuLV-mKate (half DMEM, half virus supernatant) and incubated at 37°C for 48 h. Medium was changed after 16 h of incubation. Afterward cells were analyzed via fluorescence microscopy.
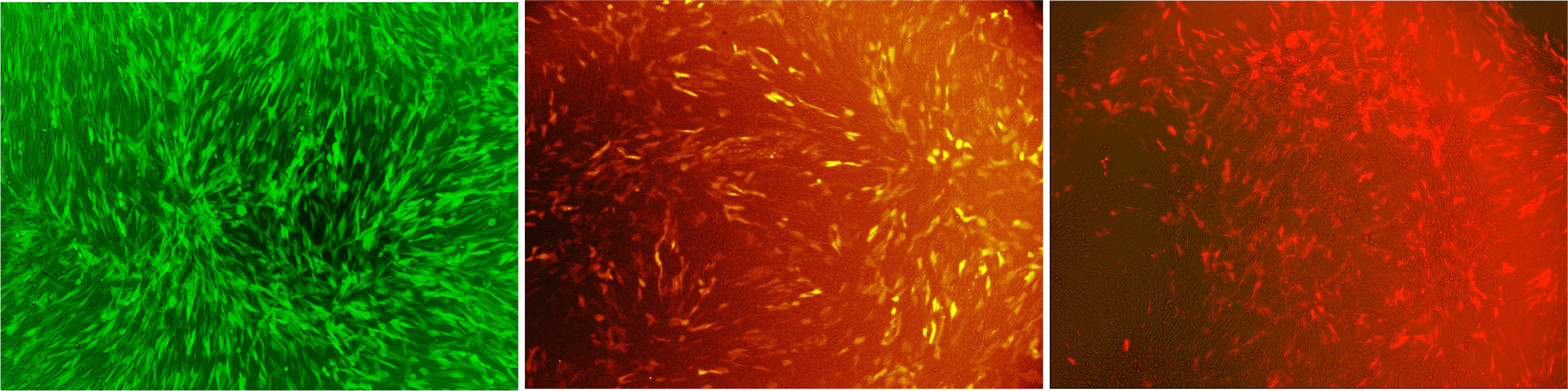
Figure 1: Murine cells infected with our viral vector
The vector contained different fluorescent markers; (A) EGFP; (B) mKO; (C) mKate that were stably integrated into the genomeLabjournal
2014/09/29
QR-code with virus on 96W plate
- Cells were transfected in suspension with the blue light system plus receptor (PKM292, PKM297, ls003, 3 x 10^5 cells/ml, 100 µl transfection mix/ml, 1.5 µg DNA/ml). Cells were incubated for 5 h at 37°C before centrifugation (900 RPM, 3 min, 24°C). Cells were resuspended with fresh DMEM to a final concentration of 1.5 x 10^5 cells/ml and seeded (0,1 ml/well).
- After 15 h of incubation cells were illuminated with blue light for 1 h.
- Cells were incubated for 24 h and afterwards infected with MuLV CMV SEAP. Medium was changed after 4 h of infection.
- After 24 h the supernatant was analyzed via SEAP assay as described before.
Viral Vectors - October
2014/10/10
Generation of QR code on 384W plates
- Cells were transfected in suspension with the blue light system using SEAP as reporter (PKM292, PKM297, PKM084, 3 x 10^5 cells/ml, 100 µl transfection mix/ml, 1.5 µg DNA/ml). Cells were incubated for 5 h at 37°C before centrifugation (900 RPM, 3 min, 24°C). Cells were resuspended with fresh DMEM to a final concentration of 2 x 10^5 cells/ml and seeded (20 µl/well).
- After 24 h of incubation cells were illuminated with blue light for 5 h.
- After 24 h the supernatant was analyzed via SEAP assay as described before.
- Wells that contained SEAP changed their colour to yellow

Figure 4: Generation of a QR code on a 384 well plate.
HEK293 cells were transfected with the blue light system (PKM292, PKM297) and light induced SEAP as reporter (PKM084). Single wells were covered with a photo mask to prevent cell from light exposure using a photo mask for a QR code (made by iGEM Team Aachen 2014). 24 hours after illumination with blue light a SEAP assay was performed leading to colour changing of wells containing SEAP. Only a part of the plate was used. (A) Photo mask for QR code generation, (B) Picture of the 384W plate after the SEAP assay, (C) the same plate with contrast of the colours increased at a computer.
Light-System
Light System - June
2014/06/16
Transfection Red light system in CHO cells
We transfected the lightsystem (pKM022) + mCherry (pKM078) & lightsystem (pkMpKM022) + receptor (pMW:SLC7A1)
- we used 4 x 100 mm dishes: 2 x for 660 nm and 2 x for 740 nm illumination (one of each for negative control)
- transfection with red light system (pKM022) and receptor (SLC7A1) or light induced mCherry (pKM078)
- transfection mix was incubated for 20 min and 1 ml added to each dish
- medium was changed after 4-5h
- cells incubated for 20 h
2014/06/17
PCB addition
- 50 mg PCB was dissolved in 2 ml DMSO - every step was performed in the dark!
- c(PCB) = 4.39x10^-5 M = 43,9 mM
- 14,08 µl PCB solution was diluted in 40 ml DMEM
- c(PCB, diluted) = 15 mM
- the culture medium of the transfected cells was replaced by 10 ml prewarmed PCB-DMEM solution
- Three plates were placed for one hour in the 660 nm lightbox (negative control, pKM078, pMW)
- Three plates were placed for one hour in the 740 nm lightbox (negative control, pKM078, pMW)
- after illuminations the lightboxes were incubated at 37 °C for 23 h
2014/06/18
Lysis of cells with RIPA buffer
- For samples transfected with the receptor cells were lysed with RIPA buffer
- Cells were washed with ice-cold PBS
- Cells were lysed with 4,43 ml ice-cold RIPA buffer (completed with phosphatase inhibitor mix) and incubated for 10 min
- Cells were detached from the plate with a inverted pipette tip and transferred into Eppis
- Eppis were vortexed and incubated on ice for 10 min
- Eppis were centrifuged for 5 min at 10.000 x g
- 60 µl direct-lysate with were mixed with 15 µl 5x SDS-Dye and heated for 10 min at 85 °C
- Lysates were stored at - 24 °C
Fixation with PFA
- Cells transfected with pKM078 (mCherry) illuminated at 740 nm, 600 nm; negative control lighted at 660 nm were washed with 30 ml ice-cold PBS
- 3 ml ice-cold PFA-Solution (4%) was added and 10 min incubated on ice
- PFA was replaced with 7 ml PBS and stored in the freezer
Experiment did not work.
2014/06/22
Transfection Red light system in CHO cells
- for each transfection condition an 1.5 ml eppi was prepared with:
- 50 µl Opti-MEM
- plasmid DNA (0.75 µg in total)
- both were mixed by vortexing
- 2.5 µl PEI solution was added
- mixture was directly vortexed for 10 sec
- the solution was incubated at room temperature for 15 min
- the precipitate (50 µl per well) was carefully added to the CHO cells
- the mixture was incubated in the cell incubator
- after 5 h: the medium was changed to prewarmed DMEM
- the CHO cells were incubated for 20 h
- 3 sample mixtures were prepared and the medium in the wells with 0.5 ml of one of the mixtures replaced:
- 10 ml DMEM - for samples 1,4,5 and 6
- 3.5 ml DMEM + 1.3 µl PCB solution - samples 3 and 7
- 3.5 ml DMEM + 1.3 µl DMSO - samples 2
- (DMSO was diluted 1:8000 in methanol:HCL (95:5 vol/vol))
- A 6W plate was prepared:
- 1st well: negative control
- 2nd well: transfection control - pEGFP_C1 (3 µg)
- 3rd well: transfection control - pEGFP_C1 (3 µg)
- 4th well: SLC7A1 (3 µg)
- 5th well: SLC7A1 (3 µg)
- 6th well: SLC7A1 (2 µg)
- cells were washed with 0.5 ml PBS
- 200 µl RIPA buffer was added to the well
- cells were scraped off and transferred into a tube
- cells were incubated on ice on 10 min
- the mix was vortexed and incubated on ice for 10 min
- lysates were centrifuged for 5 min at 10.000 x g
- 170 µl direct-lysate of each sample was added to 42.5 µl 5x SDS loading-dye
- samples were incubated for 10 min at 95°C lysates were stored at -26°C
- 2x 24 well plates were trancfected, one plate with cover slips and one plates without cover slips
- 24 well plate with cover slips:
- 5 wells: 0.5 µg pKM022 + 0.25 µg pKM078 + PCB
- 5 wells: 0.5 µg pKM022 + 0.25 µg pKM078 - pCB
- 5 wells: 0.5 µg pSAM200 + 0.25 µg pKM078
- 3 wells: 0.5 µg EGFP_C1 + 0.25 µg pKM078
- 3 wells: 0.75 µg EGFP_C1
- 3 wells: negatve control
- 24 well plate without cover slips:
- 5 wells: 0.5 µg pKM022 + 0.25 µg pKM006 + PCB
- 5 wells: 0.5 µg pKM022 + 0.25 µg pKM006 - pCB
- 5 wells: 0.5 µg pSAM200 + 0.25 µg pKM006
- 3 wells: 0.5 µg EGFP_C1 + 0.25 µg pKM006
- 3 wells: 0.75 µg EGFP_C1
- 3 wells: negatve control
- transfection mastermixes were incubated for 15 min
- after 5h: medium exchange in both 24 well plates
- after 21h: medium was changed (0.5 ml per well)
- 5 wells (0.5 µg pKM022 + 0.25 µg pKM006 + PCB): DMEM medium supplemented with 15 µM PCB
- 5 wells (0.5 µg pKM022 + 0.25 µg pKM078 + PCB): DMEM medium supplemented with 15 µM PCB
- 5 wells (0.5 µg pKM022 + 0.25 µg pKM006 - PCB): DMEM medium supplemented with DMSO
- 5 wells (0.5 µg pKM022 + 0.25 µg pKM006 - PCB): DMEM medium supplemented with DMSO
- in all other wells: medium was changed to prewarmed DMEM medium
- both 24 well plates were illuminated for 24 h in the 660 nm lightbox at 37°C
- intensity of our LED band light box: ca. 6 µmol/(m^2*s)
- intensity of both light boxes from other laboratory (AG Weber): 20 µmol/(m^2*s)
- transfection scheme in 24 well:
- medium was changed 5h after transfection
- Two solution-mixes were prepared:
- 1) 2x (15 µl our PCB solution + 37.5 ml DMEM)
- 2) 2.8 µl Weber's BCB solution + 7 ml DMEM)
- the medium in all wells was aspirated
- 0.5 ml solution-mix 1 was added to wells labeled with A, B, D, F, G, H
- 0.5 ml solution-mix 2 was added to wells labelled with C
- plates were first incubated in the dark for 1h then illuminated for 24h
- 2.25 g homoarginine + 0.048 g MgCl2 diluted in 250 ml ddH2O
- 105 ml diethanolamine was added
- pH value was set to 9.8 with HCL solution
- filled up to 500 ml with ddH2O
- 96 well plates were sealed and incubaed at 65°C for 45 min
- 1 min centrifuged at 1258 g
- 100 µl SEAP buffer was prepared in each well of a 96 (flat-bottom) well plate
- of the centrifguged plates 80 µl supernatant per well were added to the the 100 µl SEAP buffer
- each well was completed with 20 µl pNPP (substrate)
- spectroscopic measurement every minute for 1 hour at 405 nm
- medium was changed afetr 5h
- PCB was added 20h after medium exchange
- incubation for 1h in the dark
- 1st plate was incubated for 24h at 660 nm illumination in our LED band box
- 2nd plate was incubated for 24h in the dark
- 200 µl supernatant were transferred in a 96W plate for performing a SEAP assay.
- some plates were fixed with PFA for microscopy, and stained with DAPi
- pKM297: VP16-PDZb (in two fold excess tranfected)
- pKM292: Gal4BD, LOV (in two fold excess tranfected)
- pKM084: SEAP, reporter (1x transfected)
- pMIG-IRES-GFP: junk
- C1_EGFP: reansfection control
- the medium was changed 5h after trasfection (48 µl), first incubated for 20h in the dark and then incubated for 5h, 2.5h and 1h:
- 1st plate in our LED band box at 465 nm
- 2nd plate in the dark
- a SEAP assay was performed after 24 hours of incubation
- transfection of the red-light system
- 3x 24 well plates with:
- 3x 24 well plates with:
- medium was changed after 5 hours
- PCB was added after 20 hours
- mastermixes:
- 40 ml DMEM + 14.5 µl PCB
- 32 ml HTS + 11.5 µl PCN
- incubation for 1h in the dark
- for each of the 3 plates with the mCherry reporter one plate was incubated in the dark, one plate at 740 nm and one plate at 660 nm illumination for 24 hours
- for each of the 3 plates with the SEAP reporter one plate was incubated oin the dark, one at 740 nm illumination and one at 660 nm illumination for 24 hours
- 200 µl supernatant of each well were transferred into a 96W plate for performing a SEAP-assay
- the medium was changed 5 hours after trasfection and incubated for 20h in the dark
- 1st plate: incubation in our LED band box at 465 nm for 5 hours
- 2nd plate incubation in the dark
- a SEAP assay was performed 24 hours after illumination
| pKM078/pKM006 | pKM022 | pSAM200 | pMIG | Opti-MEM (to 200 µl) | PEI | ||
| 1) mCherry for leaky expression | 58 µl pKM078 | / | / | 5.2 µl | 136,8 µl | 10 µl | |
| 2) pKM078 -PCB | 58 µl | 4.5 µl | / | 1.7 µl | 135.8 µl | 10 µl | |
| 3) pKM078 +PCB | 58 µl | 4.5 µl | / | 1.7 µl | 135.8 µl | 10 µl | |
| 4) constitutive control | 58 µl | / | 19 µl | 1.7 µl | 138.4 µl | 10 µl | |
| 5) transfection control | / | / | / | 7 µl | 193 µl | 10 µl | |
| 6) SEAP for leaky expression | 1.3 µl pKM006 | / | / | 5.2 µl | 193.5 µl | 10 µl | |
| 7) pKM 006 +PCB | 1.3 µl | 4.5 µl | / | 1.7 µl | 194.2 µl | 10 µl |
2014/06/23
PCB addidtion
To activate the red light system PCB was added after 20 hours of incubation
2014/06/29
Transfection of receptor (SLC7A1) in CHO cells)
Cells were lysed with RIPA buffer 24 hours after incubation at 37°C
transfection of the red light system into CHO cells
2014/06/30
PCB addition
Cells were lysed with RIPA buffer after 24 hours. Lysates were incubated with SDS loading dye for 10 min at 95°C and stored at -24°C.
Light System - July
2014/07/07
Measurement of LED band
2014/07/08
Transfection of red light system in CHO cells/ comparison of different light boxes
| A: 0.5 µg pKM022 + 0.25 µg pKM006 | A: 0.5 µg pKM022 + 0.25 µg pKM006 | A: 0.5 µg pKM022 + 0.25 µg pKM006 | B: 0.5 µg pSAM200 + 0.25 µg pKM006 | B: 0.5 µg pSAM200 + 0.25 µg pKM006 | B: 0.5 µg pSAM200 + 0.25 µg pKM006 |
| C: 0.5 µg pKM022 + 0.25 µg pKM006 | C: 0.5 µg pKM022 + 0.25 µg pKM006 | C: 0.5 µg pKM022 + 0.25 µg pKM006 | D: 0.5 µg empty vector + 0.25 µg pKM006 | D: 0.5 µg empty vector + 0.25 µg pKM006 | D: 0.5 µg empty vector + 0.25 µg pKM006 |
| E: 0.75 µg pMIG | E: 0.75 µg pMIG | E: 0.75 µg pMIG | F: 0.5 µg pKM022 + 0.25 µg078 (on cover slides) | F: 0.5 µg pKM022 + 0.25 µg078 (on cover slides) | F: 0.5 µg pKM022 + 0.25 µg078 (on cover slides) |
| G: pHB362 + pKM082 | G: pHB362 + pKM082 | G: pHB362 + pKM082 | H: pHB362 + pKM082 | H: pHB362 + pKM082 | H: pHB362 + pKM082 |
2014/07/09
PCB addition
To activate the red light system PCB was added after 20 hours of incubation
After 24 hours of illumination 200 µl supernatant of samples A, B, C, D, E, G, H was transferred in 96W plates. The plate was stored at -24°C before measurement with SEAP-assay. Samples F were fixed with PFA and stored at 4°C.
2014/07/11
Preparation of SEAP buffer
SEAP assay Red light system/ comparison different light boxes
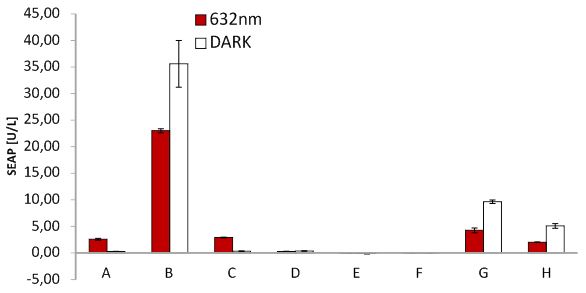
SEAP assay with illuminated CHO cells transfected with the red light system, box of our lab. A: pKM022 + pKM006; B: pSAM200 + pKM006; C: pKM022 + pKM006; D: empty vector + pKM006; E: transfection control; F: pKM022 + pKM078 (on cover slides);G: pHB362 + pKM082; H: pHB362 + pKM082
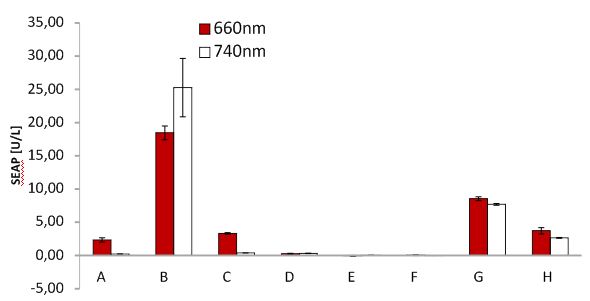
SEAP assay with illuminated CHO cells transfected with the red light system, box of other lab. A: pKM022 + pKM006; B: pSAM200 + pKM006; C: pKM022 + pKM006; D: empty vector + pKM006; E: transfection control; F: pKM022 + pKM078 (on cover slides);G: pHB362 + pKM082; H: pHB362 + pKM082

SEAP assay with illuminated CHO cells transfected with the red light system, comparison of two different light boxes. A: pKM022 + pKM006; B: pSAM200 + pKM006; C: pKM022 + pKM006; D: empty vector + pKM006; E: transfection control; F: pKM022 + pKM078 (on cover slides);G: pHB362 + pKM082; H: pHB362 + pKM082
2014/07/11
Transfection of CHO cells with the red light system.
| 1: 0.5 µg pKM022 + 0.25 µg pKM006 | 1: 0.5 µg pKM022 + 0.25 µg pKM006 | 1: 0.5 µg pKM022 + 0.25 µg pKM006 | 2: 0.5 µg pSAM200 + 0.25 µg pKM006 | 2: 0.5 µg pSAM200 + 0.25 µg pKM006 | 2: 0.5 µg pSAM200 + 0.25 µg pKM006 |
| 4: 0.75 µg pMIG-IRE-GFP | 4: 0.75 µg pMIG-IRES-GFP | 4: 0.75 µg pMIG-IRES-GFP | 3: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 | 3: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 | 3: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 |
| 5: 0.5 µg pKM022 + 0.25 µg pKM078 (on cs) | 5: 0.5 µg pKM022 + 0.25 µg pKM078 (on cs) | 5: 0.5 µg pKM022 + 0.25 µg pKM078 (on cs) | 6: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 (on cs) | 6: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 (on cs) | 6: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 (on cs) |
| 7: 0.5 µg pSAM200 + 0.25 µg pKM078 (on cs) | 7: 0.5 µg pSAM200 + 0.25 µg pKM078 (on cs) | 7: 0.5 µg pSAM200 + 0.25 µg pKM078 (on cs) | 8: 0.5 µg pKM022 + 0.25 µg pKM006 + SLC7A1 (on cs) | 8: 0.5 µg pKM022 + 0.25 µg pKM006 + SLC7A1 (on cs) | 8: 0.5 µg pKM022 + 0.25 µg pKM006 + SLC7A1 (on cs) |
CHO cells transfected with PKM022 and p14rz_002 after illumination (A); dark control (B); positiv control (PSAM200 and p14rz_002) (C)
2014/07/15
Transfection of CHO and HEK cells with the blue light system.
| pKM297 (2x) + pKM292 (2x) + pKM084 (1x) (CHO) | pKM297 (2x) + pKM292 (2x) + pKM084 (1x) (CHO) | pKM297 (2x) + pKM292 (2x) + pKM084 (1x) (CHO) | pKM297 (2x) + pKM292 (2x) + pKM084 (1x) (HEK) | pKM297 (2x) + pKM292 (2x) + pKM084 (1x) (HEK) | pKM297 (2x) + pKM292 (2x) + pKM084 (1x) (HEK) |
| pKM297 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (CHO) | pKM297 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (CHO) | pKM297 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (CHO) | pKM297 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (HEK) | pKM297 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (HEK) | pKM297 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (HEK) |
| pKM292 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (CHO) | pKM292 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (CHO) | pKM292 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (CHO) | pKM292 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (HEK) | pKM292 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (HEK) | pKM292 (2x) + pKM084 (1x) + pMIG-IRES-GFP (2x) (HEK) |
| C1_EGFP (CHO) | C1_EGFP (CHO) | C1_EGFP (CHO) | C1_EGFP (HEK) | C1_EGFP (HEK) | C1_EGFP (HEK) |

HEK cells transfected with the blue light system (PKM292, PKM297 and PKM084) and illuminated for different durations. A SEAP assay was performed afterwards.
2014/07/17
Transfection of different CHO cell lines with the red light system.
| DMEM CHO: 0.5 µg pKM022 + 0.25 µg pKM078 | DMEM CHO: 0.5 µg pKM022 + 0.25 µg pKM078 | DMEM CHO: 0.5 µg pKM022 + 0.25 µg pKM078 | DMEM CHO: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 | DMEM CHO: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 | DMEM CHO: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 |
| DMEM CHO: 0.5 µg pSAM200 + 0.25 µg pKM078 | DMEM CHO: 0.5 µg pSAM200 + 0.25 µg pKM078 | DMEM CHO: 0.5 µg pSAM200 + 0.25 µg pKM078 | DMEM CHO: 0.75 µg pMIG-IRES-GFP | DMEM CHO: 0.75 µg pMIG-IRES-GFP | DMEM CHO: negative control |
| HTS CHO-K1: 0.5 µg pKM022 + 0.25 µg pKM078 | HTS CHO-K1: 0.5 µg pKM022 + 0.25 µg pKM078 | HTS CHO-K1: 0.5 µg pKM022 + 0.25 µg pKM078 | HTS CHO-K1: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 | HTS CHO-K1: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 | HTS CHO-K1: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM078 |
| HTS CHO-K1: 0.5 µg pSAM200 + 0.25 µg pKM078 | HTS CHO-K1: 0.5 µg pSAM200 + 0.25 µg pKM078 | HTS CHO-K1: 0.5 µg pSAM200 + 0.25 µg pKM078 | HTS CHO-K1: 0.75 µg pMIG-IRES-GFP | HTS CHO-K1: 0.75 µg pMIG-IRES-GFP | HTS CHO-K1: negative control |
| DMEM CHO: 0.5 µg pKM022 + 0.25 µg pKM006 | DMEM CHO: 0.5 µg pKM022 + 0.25 µg pKM006 | DMEM CHO: 0.5 µg pKM022 + 0.25 µg pKM006 | DMEM CHO: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 | DMEM CHO: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 | DMEM CHO: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 |
| DMEM CHO: 0.5 µg pSAM200 + 0.25 µg pKM006 | DMEM CHO: 0.5 µg pSAM200 + 0.25 µg pKM006 | DMEM CHO: 0.5 µg pSAM200 + 0.25 µg pKM006 | DMEM CHO: 0.75 µg pMIG-IRES-GFP | DMEM CHO: 0.75 µg pMIG-IRES-GFP | DMEM CHO: negative control |
| HTS CHO-K1: 0.5 µg pKM022 + 0.25 µg pKM006 | HTS CHO-K1: 0.5 µg pKM022 + 0.25 µg pKM006 | HTS CHO-K1: 0.5 µg pKM022 + 0.25 µg pKM006 | HTS CHO-K1: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 | HTS CHO-K1: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 | HTS CHO-K1: 0.5 µg pMIG-IRES-GFP + 0.25 µg pKM006 |
| HTS CHO-K1: 0.5 µg pSAM200 + 0.25 µg pKM006 | HTS CHO-K1: 0.5 µg pSAM200 + 0.25 µg pKM006 | HTS CHO-K1: 0.5 µg pSAM200 + 0.25 µg pKM006 | HTS CHO-K1: 0.75 µg pMIG-IRES-GFP | HTS CHO-K1: 0.75 µg pMIG-IRES-GFP | HTS CHO-K1: negative control |

SEAP assay performed with two different CHO cell lines. CHO 2 grow in completed DMEM medium and CHO K1 in completed HTS medium.
2014/07/22
Transfection of blue light system in HEK cells
| pKM 292 + pKM 297 + pKM084 | pKM 292 + pKM 297 + pKM084 | pKM 292 + pKM 297 + pKM084 | - | - | - |
| pKM297 + pKM084 + junk | pKM297 + pKM084 + junk | pKM297 + pKM084 + junk | - | - | - |
| pKM292 + pKM084 + junk | pKM292 + pKM084 + junk | pKM292 + pKM084 + junk | - | - | - |
| pMIG_IRES_GFP | pMIG_IRES_GFP | pMIG_IRES_GFP | - | - | - |

SEAP assay performed with HEK cells transfected with the blue light system (PKM292 and PKM297) with seap as a reporter gene (PKM084).
Light System - August
2014/08/02
Pattern Generation
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
5h |
|
2 |
5h |
|
3 |
5h |
|
4 |
5h |
|
5 (dark control) |
5h |
We were not able to detect a pattern when irradiating for 5 h.
Dark control showed no fluorecent cells.
Positive control showed fluorecent cells.
2014/08/10
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
30 min |
|
2 |
1 h |
|
3 |
1/1/2 h |
|
4 |
2 h |
|
5 (dark control) |
2 h |
We were not able to detect a pattern when irradiating for the stated timepoints.
Dark control showed no fluorecent cells.
Positive control showed fluorecent cells.
2014/08/15
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
1 min |
|
2 |
10 min |
|
3 |
15 min |
|
4 |
Pulsed - 1 sec/29 sec (for 10 min) |
|
5 (dark control) |
15 min |
We detected fluorecence but no pattern.
Dark control showed fluorecent cells.
Positive control showed fluorecent cells.
2014/08/21
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
8.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
8.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
1 min |
|
2 |
10 min |
|
3 |
15 min |
|
4 |
Pulsed - 1 sec/29 sec (for 10 min) |
|
5 (dark control) |
15 min |
We detected fluorecence but no pattern.
Dark control showed fluorecent cells.
Positive control showed fluorecent cells.
2014/08/30
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
1 min |
|
2 |
10 min |
|
3 |
15 min |
|
4 |
Pulsed - 1 sec/29 sec (for 10 min) |
|
5 (dark control) |
15 min |
We detected no fluorecence.
Dark control showed no fluorecent cells.
Positive control showed fluorecent cells.
Light System - September
2014/09/04
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
1 min |
|
2 |
10 min |
|
3 |
15 min |
|
4 |
Pulsed - 1 sec/29 sec (for 10 min) |
|
5 (dark control) |
15 min |
We detected no fluorecence.
Dark control showed no fluorecent cells.
Positive control showed fluorecent cells.
2014/09/09
Kinetik of the blue light induced receptor
HEK293 cells were transfected using PEI with the blue light system (PKM292 and PKM297) and the light induced receptor (p14ls_003). 24 hours after transfection cells were illuminated for 5 hours with blue light. At distinct time points (after 10h, 12h, 15h, 18h and 24h) cells were analysed with fluorescence microscopy and lysed with RIPA buffer for analysis with Western blot.

Kinetik of the blue light induced receptor.
HEK cells were transfected with the blue light system (PKM292 and PKM297) and the light induced receptor (p14ls_003, mCherry labeled receptor). Pictures were taken after 12h, 15h, 18h and 24h.
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
1 min |
|
2 |
10 min |
|
3 |
15 min |
|
4 |
Pulsed - 1 sec/29 sec (for 10 min) |
|
5 (dark control) |
15 min |
We detected no fluorecence.
Dark control showed no fluorecent cells.
Positive control showed fluorecent cells.
2014/09/22
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
30 min |
|
2 |
1 h |
|
3 |
1/1/2 h |
|
4 |
2 h |
|
5 (dark control) |
2 h |
We detected fluorecence in all experiments.
2014/09/26
Pattern generation with blue light system
Transfecting HEK293T cells with:
all plasmids in a concentration of 500 µg/µl
positive control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
14.4 |
336.0 |
|
pKM292 |
- |
||
|
pEZ422 |
4.8 |
||
|
Ls003 |
4.8 |
negative control (1 x 6 cm plate):
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
- |
- |
- |
|
pKM292 |
- |
||
|
pEZ422 |
- |
||
|
Ls003 |
- |
experiment (5 x 6 cm plates)
|
Construct |
DNA [µl] |
PEI [µl] |
OptiMEM [µl] |
|
pKM297 |
16.0 |
72.0 |
1680.0 |
|
pKM292 |
16.0 |
||
|
pEZ422 |
- |
||
|
Ls003 |
16.0 |
Medium was changed 5 h post transfection
All further steps were performed under dark conditions to prevent accidental activation of the light system.
Plates were put into light-boxes 24 h post transfection
Plates were irradiated for:
|
Plate |
Duration |
|
1 |
1 min |
|
2 |
5 min |
|
3 |
10 min |
|
4 |
15 min |
|
5 (dark control) |
15 min |
We detected fluorecence in all experiments.

The light induced receptor was induced by light scatter.
HEK293T cells were transfected with the light system and the light induced receptor (p14ls_003) labeled with mCherry. Cells were illuminated using a photo mask that coverd parts of the cell culture and prevented them from light exposure. However, the receptor expression was activated.
Light System - October
Standardization
Standardization - August
PCR mix for site-directed mutagenisis (25 µl)
| template | 1 µl |
| buffer (5x) | 5 µl |
| Primer | 1 µl each |
| polymerase | 0.5 µL |
| dNTPs | 1 µl |
| DMSO | 0.5 µl |
| water | 15 µl |
start your PCR mix with just one primer and let reaction run for ten cycles including end elongation. shortly after that add 1 µl dNTP and 1 µl of primer 2. start second PCR under same conditions for 15 cycles. Don't spend too much time with adding dNTPs and primer 2.
DNA amplifications such for ligation PCR approaches contained 50 µl.
protocoll site-directed mutagenisis
| 98 °C | 5 min | denaturation |
| 98 °C | 30 s | denaturation |
| 60 °C | 30 s | annealing |
| 72 °C | 30 s/kb | elongation |
| 72 °C | 10 min | end elongation |
To avoid high background activities by not-mutated plasmids, the PCR mixture was digested by DpnI. It cuts methylated DNA only, leading to remove the DNA template.
Therefore transformation is done with unmethylated PCR product, where plasmids contain the desired mutation.
DpnI digest
add 3 µl Cutsmart, 0.5 µl water and 0.5 µl DpnI. Incubate at 37 °C for 90 minutes, followed by an inactivation step at 80 °C for 20 minutes.
test digest
PCR products or test digests of plasmids where separated gelelectrophoresis, containing 1 % agarose. Small gels were run at 95 V for 90 minutes, large gels at 105 V for 90 minutes.
2014.08.21.
| PCR # | template | primer 1 | primer 2 | polymerase | product | size | annealing temperature | elongation time |
| 1 | pKM297 | o14_sd_003 | o14_sd_004 | Q5 | ePDZb_G1448C_PstI | 3.8 kb | 60 °C | 01:55 min |
| 2 | pKM292 | o14_sd_011 | o14_sd_012 | Q5 | GAL4_binding_domain_RFC_25 | 0.5 kb | 60 °C | 00:16 min |
| 3 | pKM292 | o14_sd_019 | o14_sd_020 | Q5 | LOV_C3423T_PstI | 3.7 kb | 60 | 01:53 min |
| 4 | zl_003 | o14_sd_023 | o14_sd_024 | Q5 | P2A_RFC_10 | 0.1 kb | 60 | 00:04 min |
| 5 | zl_003 | o14_sd_027 | o14_sd_028 | Q5 | P2A_C9374G_NgoMIV | 11.7 kb | 60 | 05:50 min |
Each PCR was done in triplicates and digested with DpnI. Each replicate was used for transformation with competent bacteria on a seperated Ampicillin plate and incubated at 37 °C. bacteria containing zl_003 were incubated at 32 °C.
2014.08.22.
| PCR # | template | primer 1 | primer 2 | polymerase | product | size | annealing temperature | elongation time |
| 6 | zl_oo3 | o14_sd_029 | o14_sd_030 | Phusion | puromycin_resistance_gene_RFC_25 | 0.6 kb | 60 °C | 00:20 min |
| 7 | rz_008 | o14_sd_008 | o14_sd_009 | Phusion | CAT-1_C1168T_PstI | 6.4 kb | 60 °C | 03:15 min |
| 8 | pKM084 | o14_sd_047 | o14_sd_048 | Phusion | SEAP_G1419C_PstI | 5.4 kb | 60 | 02:45 min |
All PCRs were digested with DpnI and transformed on Ampicillin containing plates at 37 °C.
Transformation from PCR 1.1/1.2/1.3, 3.1/3.2/3.3, 5.1/5.3 were successful, but entirely PCR 2 & 4 not (repeated with inkubation at 32 °C). Three colonies were picked from each plate and used for over night culures (ONC).
Advice from instructor: an elongation time of 30 s for each DNA fragment smaller than 1 kb will be used.
2014.08.23.
A mini prep kit from Quiagen (250 reactions) were used for DNA isolation:
| template | concentration [ng/µl] | template | concentration [ng/µl] |
| 1.1 I | 155.2 ng/µl | 1.2 I | 174.0 ng/µl |
| 1.1 II | 153.1 ng/µl | 1.2 II | 727.5 ng/µl |
| 1.1 III | 566.6 ng/µl | 1.2 III | 562.5 ng/µl |
| 1.3 I | 265.8 ng/µl | 3.1 I | 174.1 ng/µl |
| 1.3 II | 226.4 ng/µl | 3.1 II | 144.5 ng/µl |
| 1.3 III | 187.7 ng/µl | 3.1 III | 144.4 ng/µl |
| 3.2 I | 197.0 ng/µl | 3.3 I | 163.1 ng/µl |
| 3.3 II | 163.5 ng/µl | 5.1 I | 150.0 ng/µl |
| 5.3 I | 448.7 ng/µl | 5.1 II | 442.2 ng/µl |
| 5.3 II | 230.1 ng/µl | 5.1 III | 295.7 ng/µl |
| 5.3 III | 559.4 ng/µl |
test digest with:
| PCR | enzyme | buffer | fragment size |
| 1 | PstI/ClaI | CutSmart | mutated: 2.4 kb, 1.5 kb, not mutated: 2.4 kb, 0.9 kb, 0.6 kb |
| 3 | PstI/BbsI | CutSmart | mutated: 2.4 kb, 0.65 kb, 0.53 kb; not mutated: 2.4 kb, 0.53 kb, .04 kb, .026 kb, 0.23 kb, 0.06 kb |
| 5 | AgeI/NgoMIV | NEB 1.1 | mutated: 5.1 kb, 4.7 kb, 1.8 kb; not mutated: 4.7 kb, 4.1 kb, 2.8 kb |
0.625 µl BbsI was added, due to its decreased activity (75%) in NEB 1.1. Test digests were done over night.
Transformations of PCR 8 and PCR 5 didn't work. Repeated in duplicates: SEAP was incubated at 37 °C, P2A at 32 °C.
5 ONC of PCR 7 (CAT-1) were done.
2014.08.24.
results of gel electrophoresis:

gel electrophoresis of PCR 1 & 5.
PCR 1: PCR 1.1 I and PCR 1.1 II showed perfect fragment sizes (2.3 kb & 1.4 kb). àCandidates for sequencing.
PCR 5: PCR 5.1 & PCR 5.3 were successful. DNA fragments at 5.1 kb and 4.7 kb. Fragment at 2 kb invisible, but if DNA wasn’t mutated, there would not be a fragment at 5.1 kb. à Candidates for sequencing.

gel electrophoresis of PCR 3.
PCR 3: PCR 3.1 III looked fine. Fragments as expected.à Candidate for sequencing.
Despite missing sequencing results, PCR 5.1 I was used for a transformation.
DNA extraction of CAT-1 (5x)
|
PCR # |
Concentration [ng/µl] |
|
7 I |
489.2 |
|
7 II |
316.5 |
|
7 III |
385.0 |
|
7 IV |
451.8 |
|
7 V |
550.0 |
Test digest with:
|
PCR # |
enzyme |
buffer |
fragment size |
|
7 |
AgeI/PstI-HF |
CutSmart |
mutated:4.2 kb, 1.6 kb not mutated: 4.2 kb, 1.3 kb, 0.3 kb |
test digest was done overnight.
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
10 |
pKM292 |
o14sd_011 |
014sd_012 |
Phusion |
GAL4_RFC_25 |
0.5 kb |
55 °C |
|
11 |
zl_003 |
o14sd_29 |
014sd_030 |
Phusion |
Puromycin_resistance_gene_RFC_25 |
0.6 kb |
55 °C |
2014.08.25
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
12 |
pKM084 |
o14sd_047 |
014sd_048 |
Phusion |
SEAP_G1419C_PstI |
5.4 kb |
55 °C |
|
13 |
zl_003 |
o14sd_055 |
014sd_056 |
Phusion |
WPRE_C530A_NgoMIV |
11.7 kb |
55 °C |
|
14 |
LOV PCR Product |
o14sd_011 |
014sd_012 |
Phusion |
GAL4_ RFC_25 |
0.5 kb |
55 °C |
Result gel electrophoresis (digest CAT-1)

gel electrophoresis of PCR 7.
Result of colony 1 looked fine. à Candidate for sequencing.
Transformation of PCR 6 & 8 did not work.
2014.08.26
DNA extraction from PCR 5.1 (5x)
|
PCR # |
Concentration [ng/µl] |
|
5.1 I |
213.0 |
|
5.1 II |
283.2 |
|
5.1 III |
254.1 |
|
5.1 IV |
198.4 |
|
5.1 V |
232.2 |
Test digests according to 2014.08.23 overnight.
2014.08.27
Gel electrophoresis from test digest PCR 5.1

gel electrophoresis of PCR 5.1.
Test digests looked fine. Fragments as expected at 5.1 kb, 4.7 kb and about 2.0 kb. PCR 5.1 I was used as a template to amplify P2A with RFC 25 prefix and suffix.
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
15 |
PCR 5.1 I |
o14sd_025 |
014sd_026 |
Phusion |
P2A_RFC_25 |
0.1 kb |
55 °C |
PCR 15 was done in 50 µl approach and triplicates.
Gel electrophoresis from PCR 15

gel electrophoresis of PCR 15.
Gel contains 2 % agarose. Marker is 100 bp from Promega. Fragments lay between 100 and 200 bp. Theoretical size 136 bp. àFragments cut out and prepared for gel extraction. All three fragments were run over one column.
PCR 14 repeated as PCR 16 due to contaminated water.
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
16 |
LOV PCR Product |
o14sd_011 |
014sd_012 |
Phusion |
GAL4_ RFC_25 |
0.5 kb |
55 °C |
Gel electrophoresis from PCR 16

gel electrophoresis of PCR 16.
Gel contains 2 % agarose. Marker is 100 bp from Promega. Fragments lay at 500 bp, expected size: 517 bp. Fragments were cut out and prepared for gel extraction. All three fragments were run over one column.
Gel extraction from P2A RFC 25 and GAL4 RFC 25
|
name |
concentration [ng/µl] |
|
GAL4 RFC 25 |
38.5 |
|
P2A RFC 25 |
34.4 |
Ligation of P2A RFC 25 and GAL4 RFC 25 in pSB1C3
Preparative digest according to test digest protocol, but incubated for four hours. Inserts and backbone were cut with EcoRI-HF/PstI-HF in CutSmart.
Used ratio backbone:insert = 1:3
Used volumes of insert and backbone
|
GAL4 RFC 25 |
1.72 µl |
pSB1C3 |
0.48 µl |
|
P2A RFC 25 |
1.7 µl |
pSB1C3 |
0.5 µl |
|
control (water) |
1.7 µl |
pSB1C3 |
0.5 µl |
Ligation was performed with T4 ligase (40.000 U/ml) from NEB
After transformed with DNA bacteria culture were incubated at 37 °C for one hour. Afterwards cultures were streaked out onto agar plates containing Chloramphenicol.
Despite missing sequencing results, ONC were made from PCR 1.1 I & PCR 3.1 III (5x each).
2014.08.28
Sequencing results: P2A not mutated, CAT-1: fail. nucleotide length 1 nt., LOV: not been uploaded.
DNA extraction from ONC PCR 1.1 I (ePDZb) & PCR 3.1 III (LOV)
|
template |
concentration [ng/µl] |
template |
concentration [ng/µl] |
|
LOV I |
103.7 |
ePDZb I |
136.8 |
|
LOV II |
91.4 |
ePDZb II |
121.4 |
|
LOV III |
119.4 |
ePDZb II |
137.0 |
|
LOV IV |
113.4 |
ePDZb IV |
102.2 |
|
LOV V |
129.9 |
ePDZb V |
155.2 |
Test digests PCR 1.1 I & PCR 3.1 III:
|
template |
enzyme |
buffer |
fragment size |
|
LOV |
XhoI/PstI-HF |
CutSmart |
mutated:2.7 kb, 0.7 kb not mutated: 2.7 kb, 0.4 kb, 0.3 kb |
|
ePDZb |
ClaI/PstI-HF |
CutSmart |
mutated:2.3 kb, 1.5 kb not mutated: 2.3 kb, 0.9 kb, 0.6 kb |

gel electrophoresis of LOV & ePDZb.
LOV didn’t work -> no fragment at 0.7 kb. ePDZb looked fine. Visible fragment at 1.5 kb as expected.
Ligation of P2A RFC 25 and GAL4 RFC 25 didn’t work. Next time preparative digest will be desalted by PCR purification kit.
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
17 |
zl_003 |
o14sd_023 |
014sd_024 |
Q5 |
P2A_RFC_10 |
0.1 kb |
55 °C |
|
18 |
zl_003 |
o14sd_057 |
014sd_058 |
Q5 |
WPRE_RFC_10 |
0.6 kb |
55 °C |
|
19 |
PCR 5.1 II |
o14sd_039 |
014sd_040 |
Q5 |
CAT-1_RFC_10 |
1.9 kb |
50 °C |
|
20 |
ePDZb I |
o14sd_005 |
014sd_006 |
Q5 |
ePDZb_A1775G_EcoRI |
3.8 kb |
50 °C |
|
21 |
pKM084 |
o14sd_047 |
014sd_048 |
Q5 |
SEAP_G1419C_PstI |
5.4 kb |
50 °C |
PCR 20 & 21 digested by DpnI and afterwards transformed onto Ampicillin plates and incubates at 37 °C
PCR 17 -19 were directly purified preparedly digested overnight with EcoRI-HF/PstI-HF in Cutsmart.
Apporach: 28 µl eluate
1 µl enzyme each
4 µl CutSmart
6 µl water
2014.08.30
ONC from GAL4 RFC 25 in pSB1C3
A Chloramphenicol plate from GAL4 RFC 25 ligation (2014.08.27.) was found in the 37 °C incubator. One colony was visible. à Three ONC were picked from this colony.
ONC from PCR 20 & 21

gel electrophoresis of PCR 17 & 18.
Plates were settled with bacteria. Three ONC were picked from each plate.
P2A looked fine. The expected size was 134 bp. Fragments were above the100 bp lane and cut out for gel extraction. WPRE showed expected lane sizes (646 bp.). Cut out for gel extraction.

gel electrophoresis of PCR 19.
Instead of 2-logfrom NEB, Promega’s 100 bp ladder was used. Expected size for CAT-1 RFC 10 were 1577 bp. The highest lane of DNA ladder is 1.5 kb -> PCR products were cut out for gel extraction.
All triplicates of one sample were run over one column.
Gel extraction from preparative digests (2014.08.29.)
|
name |
concentration [ng/µl] |
|
P2A RFC 10 (PCR 17) |
22.9 |
|
WPRE RFC 10 (PCR 18) |
53.9 |
|
CAT-1 RFC 10 (PCR 19) |
17.4 |
Ligation of WPRE RFC 10, CAT-1 RFC 10, P2A RFC 10 in pSB1C3
ratio backbone:insert = 1:3
|
|
Volume [µl] |
Volume backbone [µl] |
|
P2A |
1.68 |
0.52 |
|
CAT-1 |
1.64 |
0.56 |
|
WPRE |
1.83 |
0.37 |
|
control (water) |
1.72 |
0.48 |
We got a new ligation buffer from our instructor à Quick ligation buffer. Used buffer volume: 2.6 µl, 2.4 ligation volume and 0.2 T4 ligase (400.000 U/ml) from NEB.
5 µl ligation approaches were used for transformation. 200 µl antibiotic free LB media were added and the reaction tube was incubated at 37 °C with 400 rpm for one hour. Afterwards whole cultures were streaked out onto plates containing Chloramphenicol.
2014.08.31
DNA extraction from GAL4 RFC 25 ONC (1x), PCR 20 (3x), PCR 21 (3x)
|
template |
concentration [ng/µl] |
template |
concentration [ng/µl] |
|
GAL4 RFC 25 in pSB1C3 |
154.2 |
(PCR 21) SEAP I |
188.3 |
|
(PCR 20) ePDZb I |
91.4 |
(PCR 21) SEAP II |
228.3 |
|
(PCR 20) ePDZb II |
119.4 |
(PCR 21) SEAP III |
209.6 |
|
(PCR 20) ePDZb III |
113.4 |
|
|
Test digests
|
template |
enzyme |
buffer |
fragment size |
|
GAL4 RFC 25 in pSB1C3 |
EcoRI-HF/PstI-HF |
CutSmart |
2.0 kb, 0.5 kb |
|
ePDZb |
EcoRI-HF/PstI-HF/NotI-HF |
CutSmart |
mutated:1.0 kb not mutated: 0.6 kb, 0.4 kb |
|
SEAP |
PstI-HF/BbsI |
CutSmart |
mutated: 4.1 kb, 1.3 kb not mutated: 4.1 kb, 1 kb, 0.3 kb |
Digests pipetted according to protocol.

gel electrophoresis of PCR 21.
Digests of GAL4 and ePDZb were run over 2 % agarose gel. Promega 100 bp was used as DNA ladder. SEAP was run over normal gel, with standard DNA ladder.
Fragment sizes weren’t as expected. Five new colonies for ONC were picked from same plate.
Standardization - September
2014.09.01
No ligation worked. They were repeated with a different protocol.
|
WPRE RFC 10 |
0.83 µl |
|
SLC7A1 RFC 10 |
7.98 µl |
|
P2A RFC 10 |
0.34 µl |
|
pSB1C3 |
always 2.75 µl |
Protocol: 2 µl 10x T4 buffer
0.2 µl T4 ligase (4e5 U/ml)
add water up to 20 µl
incubate at room temperature for 15 minutes
Transformed bacteria were streaked out onto plates containing Chloramphenicol and incubated at 37 °C
DNA extraction from SEAP ONC (5x)
|
template |
concentration [ng/µl] |
|
SEAP I |
215.7 |
|
SEAP II |
257.1 |
|
SEAP III |
285.0 |
|
SEAP IV |
139.9 |
|
SEAP V |
367.6 |
Afterwards DNA was digested according to protocol.
|
template |
enzyme |
buffer |
fragment size |
|
SEAP |
PstI-HF/NotI-HF |
CutSmart |
mutated: 3.7 kb, 1.7 kb not mutated: 3.7 kb, 1.4 kb, 0.3 kb |

test digest of overnight culture. Took from agar plate containig PCR 21.
Gel looked fine. SEAP III and SEAP IV will be sequenced. DNA Mini of CAT-1 (2014.08.22.) will be sequenced too, using another sequencing primer.
2014.09.02
Sequencing results of CAT-1 showed that it didn’t contain any PstI site anymore.
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
22 |
ePDZb_mutated |
o14sd_010 |
014sd_020 |
Phusion |
ePDZb_RFC_25_fwd -> AgeI site |
0.5 kb |
55 °C |
|
23 |
ePDZb_mutated |
o14sd_002 |
014sd_008 |
Phusion |
ePDZb_AgeI -> ePDZb_RFC_25_rev |
0.2 kb |
55 °C |
|
24 |
SEAP III |
o14sd_049 |
014sd_050 |
Phusion |
SEAP_RFC_10 |
1.6 kb |
50 °C |
|
25 |
SEAP III |
o14sd_045 |
014sd_046 |
Phusion |
SEAP_C2784G_NgoMIV |
5.4 kb |
50 °C |

test digest of PCR 22 & 23.
PCR 22 + PCR 23 were done in duplicates, PCR 24 in triplicates. Gel electrophoresis was done in 2 % gel.
All PCR but PCR 23 didn’t show expected size. PCR 23’s DNA fragments were cut out and prepared for gel extraction.
Ligation of CAT-1 and P2A in pSB1C3 didn’t work, but plate with bacteria containing WPRE RFC 10 in pSB1C3 shows eight colonies. They were picked for ONC.
Ligation of CAT-1 and P2a were repeated with a modified yesterday’s protocol. 2 µl of T4 ligase (40,000 U/ml) instead 0.2 µl T4 ligase (400,000 U/ml) were used.
2014.09.03
DNA extraction of WPRE ONC (8x)
|
template |
concentration [ng/µl] |
template |
concentration [ng/µl] |
|
WPRE RFC 10 I |
132.9 |
WPRE RFC 10 V |
130.0 |
|
WPRE RFC 10 II |
148.7 |
WPRE RFC 10 VI |
106.7 |
|
WPRE RFC 10 III |
91.9 |
WPRE RFC 10 VII |
89.0 |
|
WPRE RFC 10 IV |
148.8 |
WPRE RFC 10 VIII |
106.7 |
Test digest of WPRE RFC 10
|
template |
enzyme |
buffer |
fragment size |
|
WPRE |
PstI-HF/EcoRI-HF |
CutSmart |
mutated: 2.0 kb, 0.6 kb |

test digest of ligated WPRE RFC 10 into pSB1C3.
Digests were run over 1 % agarose gel
All eight test digests looked fine. WPRE RFC V will be sequenced. Other ligations didn’t work perhaps of damaged DNA.
Bacteria were transformed on Ampicillin plate with PCR 25.
2014.09.04
Sequencing results showed that WPRE was correctly ligated into pSB1C3.
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
26 |
CAT-1 mutated |
o14sd_031 |
014sd_032 |
Q5 |
CAT-1_C1184A_NgoMIV |
5.8 kb |
55 °C |
|
27 |
WPRE RFC 10 V |
o14sd_055 |
014sd_056 |
Q5 |
WPRE_C530A_NgoMIV |
2.5 kb |
55 °C |
|
28 |
LOV_mutated |
o14sd_021 |
014sd_022 |
Q5 |
LOV_RFC_25 |
3.8 kb |
55 °C |
|
29 |
ePDZb_EcoRI_out |
o14sd_021 |
014sd_022 |
Q5 |
ePDZb_RFC_10 |
3.8 kb |
55 °C |
|
30 |
pKM292 |
o14sd_011 |
014sd_012 |
Q5 |
GAL4 RFC 25 |
0.6 kb |
50 °C |
All PCRs were used to transform bacteria (Ampicillin, but PCR 27 Chloramphenicol) and afterwards incubated at 37 °C.
Triplicates of PCR 30 were made (50 µl) and preparative digest (EcoRI-HF/PstI-HF) was done directly in PCR tube. and afterwards purified. Concentration: 130 ng/µl.
Eight colonies were picked from plate containing PCR 15.
2014.09.05
DNA extraction of ONC PCR 15 (8x) and PCR 25 (5x)
|
template |
concentration [ng/µl] |
template |
concentration [ng/µl] |
|
P2A I |
275.4 |
SEAP I |
248.1 |
|
P2A II |
192.4 |
SEAP II |
127.1 |
|
P2A III |
176.3 |
SEAP III |
218.2 |
|
P2A IV |
339.3 |
SEAP IV |
145.9 |
|
P2A V |
373.3 |
SEAP V |
167.4 |
|
P2A VI |
304.2 |
||
|
P2A VII |
167.7 |
||
|
P2A VIII |
305.7 |
Test digests PCR 15 and PCR 25
|
template |
enzyme |
buffer |
fragment size |
|
P2A |
NgoMIV |
CutSmart |
mutated: 7.0 kb, 4.7 kb not mutated: 4.7 kb, 4.1 kb, 2.8 kb |
|
SEAP |
NgoMIV |
CutSmart |
mutated: 5,4 kb not mutated: 4.4 kb, 1 kb |

test digest of PCR 15.
P2A and SEAP separated by gel electrophoresis.

test digest of PCR 25.
Test digests of SEAP didn’t work. P2A V + VIII showed fragments with correct size.
Transformations and ligations
No transformation worked.
Following transformations were made:
PCR 26 -29, on Ampicillin plates and incubated at 37 °C
Following ligations were made:
CAT-1 RFC 10, P2A RFC 10, GAL4 RFC 25 in pSB1C3.
2014.09.06
No ligation worked.
2014.09.07
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
31 |
SEAP_PstI_out |
o14sd_049 |
014sd_050 |
Q5 |
SEAP RFC 10 |
1.6 kb |
50 °C |
|
32 |
CAT-1 mutated V |
o14sd_031 |
014sd_032 |
Q5 |
CAT-1 RFC 10 |
1.9 kb |
50 °C |
|
33 |
P2A_mutated_V |
o14sd_025 |
014sd_026 |
Q5 |
P2A RFC 25 |
0.1 kb |
55 °C |
|
34 |
GAL4_binding_domain |
o14sd_011 |
014sd_012 |
Q5 |
GAL4 RFC 25 |
0.6 kb |
55 °C |
|
35 |
WPRE RFC 10 |
o14sd_055 |
014sd_056 |
Q5 |
WPRE RFC 25 |
0.6 kb |
55 °C |
PCR 31-34 were made in 50 µl, PCR 35 in 25 µl.

test digest of PCR 31 & 32.
SEAP looked fine, but CAT-1 showed unspecific PCR products.
Gel extraction of GAL4 binding domain, SEAP, P2A
|
template |
concentration [ng/µl] |
|
GAL 4 binding domain RFC 25 |
60.8 |
|
SEAP RFC 10 |
27.9 |
|
P2A RFC 10 |
11.9 |
Ligation protocol
|
plasmid |
backbone |
P2A |
SEAP |
GAL 4 |
|
length (bp) |
2070 |
136 |
1577 |
507 |
|
concentration [ng/µl] |
27.3 |
11.9 |
27.9 |
60.8 |
|
volume [µl] |
1.83 |
0.83 |
4.10 |
0.60 |
|
water |
|
14.83 |
11.57 |
15.06 |
|
plus 2 µl 1x T4 buffer and 0.5 µl T4 ligase (400,000 U/ml) each |
||||
Approaches were incubated at room temperature for 30 minutes.
Transformation: lent cells from our instructor and added 10 µl of ligation approach and 3 µl of PCR 38 respectively.
Transformations were made in duplicates.
2014.09.08
All ligations worked. Three ONC were made from GAL4 RFC 25, P2A RFC 25, SEAP RFC 10, six from PCR 25, eight from WPRE RFC 25.
|
PCR # |
template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
36 |
CAT-1_PstI_out |
o14sd_039 |
014sd_040 |
Q5 |
CAT-1 RFC 10 |
1.9 kb |
55 °C |
|
37 |
ePDZb II (145.3 ng/µl) |
o14sd_005 |
014sd_006 |
Q5 |
ePDZB_A1775G_EcoRI |
3.8 kb |
55 °C |
PCR according to protocol, afterwards PCRs were digested by DpnI and transformed onto agar plates containing Ampicilin.
2014.09.09
DNA extractions failed due to old buffers. Agar plates were overgrown. Bacteria were diluted onto another plate. Transformation of PCR 36 failed. Repeated it with different primers
|
PCR # |
Template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
38 |
CAT-1_PstI_out |
o14sd_039 |
014sd_040 |
Q5 |
CAT-1 RFC 10 |
1.9 kb |
50 °C |
|
39 |
CAT-1_PstI_out |
o14sd_039 |
014sd_040 |
Q5 |
CAT-1 RFC 10 |
1.9 kb |
55 °C |
2014.09.10

test digest of PCR 38 & 39.
Amplifications of CAT-1 RFC 10 were successful. The amount of PCR product at 55 °C was much higher than at 50 °C. Both lanes were cut out used for ligation into pSB1C3.
|
PCR # |
Template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
40 |
CAT-1_PstI_out |
o14sd_031 |
014sd_032 |
Q5 |
CAT-1_C1184A_NgoMIV |
5.8 kb |
55 °C |
PCR according to protocol, afterwards PCR was digested by DpnI and transformed onto agar plates containing Ampicilin.
Gel extraction of CAT-1
Concentration: 21.4 ng/µl
Ligation approach:
|
Plasmid |
backbone |
CAT-1 RFC 10 |
|
length (bp) |
2070 |
1925 |
|
concentration [ng/µl] |
5.1 |
21.4 |
|
volume [µl] |
9.8 |
6.33 |
|
Water |
11.2 |
4.37 |
|
plus 2 µl 1x T4 buffer and 0.5 µl T4 ligase (400,000 U/ml) each |
Control and ligation were incubated at room temperature for 30 minutes. Transformation was done with 10 µl each. Bacteria cultures were incubated at 37 °C and 600 rpm for 60 minutes.
Transformation was done with 100, 50, 20, 10 µl to discover the optimal transformation volume.
Several ONC were made (PCR 31 – 35 (3x each), PCR 37 (10x)).
2014.09.11
DNA extraction from ONC
|
Template |
concentration [ng/µl] |
template |
concentration [ng/µl] |
|
ePDZB_A1775G_EcoRI I |
98.5 |
ePDZB_A1775G_EcoRI VI |
142.1 |
|
ePDZB_A1775G_EcoRI II |
91.5 |
ePDZB_A1775G_EcoRI VII |
78.1 |
|
ePDZB_A1775G_EcoRI III |
95.5 |
ePDZB_A1775G_EcoRI VIII |
64.0 |
|
ePDZB_A1775G_EcoRI IV |
93.8 |
ePDZB_A1775G_EcoRI IX |
80.3 |
|
ePDZB_A1775G_EcoRI V |
78.0 |
ePDZB_A1775G_EcoRI X |
71.1 |
|
GAL4 RFC 25 I |
105.9 |
WPRE_C530A_NgoMIV I |
109.3 |
|
GAL4 RFC 25 II |
123.8 |
WPRE_C530A_NgoMIV II |
119.0 |
|
GAL4 RFC 25 III |
117.6 |
WPRE_C530A_NgoMIV III |
209.5 |
|
P2A RFC 10 I |
107.3 |
SEAP RFC 10 I |
117.5 |
|
P2A RFC 10 II |
117.8 |
SEAP RFC 10 II |
123.4 |
|
P2A RFC 10 III |
113.1 |
SEAP RFC 10 III |
119.6 |
Test digests
|
Template |
enzyme |
buffer |
fragment size |
|
P2A RFC 10 |
EcoRI-HF/PstI-HF |
CutSmart |
2.0 kb,0.1 kb kb |
|
GAL4 RFC 25 |
EcoRI-HF/PstI-HF |
CutSmart |
2.0 kb, 0.5 kb |
|
SEAP RFC 10 |
EcoRI-HF/PstI-HF |
CutSmart |
2.0 kb, 1.5 kb |
|
ePDZb |
EcoRI-HF/NcoI |
CutSmart |
mutated: 3.8 kb not mutated: 3.2 kb, 0.6 kb |
Gel result

test digest of PCR 31 & 32.
It looked like ePDZb hadn’t been totally digested. The second lane lay short under 1 kb lane and not at 600 bp. WPRE RFC 10, SEAP RFC 10 and P2A didn’t show expected results. Especially it seemed that SEAP RFC 10 was actually identical with GAL 4 RFC 25 due to the lanes at 500 bp, which were expected for GAL4 RFC 25.
As a result GAL4 RFC I will be sequenced
Eight colonies from containing PCR 40 were picked for ONC.
2014.09.12
Despite to bad results on gel electrophoresis, P2A I and SEAP RFC 10 I will be sequenced.
DNA extraction from ONC
|
Template |
concentration [ng/µl] |
template |
concentration [ng/µl] |
|
CAT-1_C1184A_NgoMIV I |
473.8 |
CAT-1_C1184A_NgoMIV V |
269.1 |
|
CAT-1_C1184A_NgoMIV II |
461.6 |
CAT-1_C1184A_NgoMIV VI |
329.3 |
|
CAT-1_C1184A_NgoMIV III |
373.3 |
CAT-1_C1184A_NgoMIV VII |
452.2 |
|
CAT-1_C1184A_NgoMIV IV |
469.1 |
CAT-1_C1184A_NgoMIV VIII |
398.4 |
Test digest
|
Template |
enzyme |
buffer |
fragment size |
|
CAT-1_C1184A_NgoMIV |
XbaI/NgoMIV |
CutSmart |
mutated: 4.2, 1.2 kb,0.3 kb kb not mutated: 4.2, 1.2 kb,165 bp, 142 bp. |
Gel electrophoresis
test digest of PCR 38 & 39.
Samples were run in a 2 % agarose gel. Marker is 100 bp from Promega. Due to missing lane at 300 bp, the restriction site was still in CAT-1.
Ligation of SEAP RFC 10 and CAT-1 were repeated and digested with EcoRI-HF/PstI-HF in CutSmart buffer. Afterwards samples were purified by PCR purification Kit. Concentrations:
|
Plasmid |
backbone |
SEAP |
CAT-1 |
|
|
length (bp) |
2070 |
1577 |
1869 |
|
|
concentration [ng/µl] |
16.2 |
39.5 |
21.4 |
|
|
volume [µl] |
1.83 |
2.79 |
6.33 |
|
|
Water |
|
11.20 |
7.59 |
|
|
plus 2 µl 1x T4 buffer and 0.5 µl T4 ligase (400,000 U/ml) each |
|
|||
Transformed and incubated in Chloramphenicol containing LB-medium. 10, 50 and 100 µl of cluture were streaked out onto agar plates.
2014.09.13
Only few colonies were grown. Wait another day before using them for ONC.
Our instructor recommended us to use Pfu-Polymerase instead of Q5 or Phusion for site-directed mutagenisis PCRs.
|
PCR # |
Template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
40 |
SEAP_PstI_out (09.01.) |
o14sd_045 |
014sd_046 |
Pfu |
SEAP_G2780C_NgoMIV |
5.8 kb |
55 °C |
|
41 |
LOV_09_10 |
o14sd_039 |
014sd_040 |
Pfu |
LOV_without_PstI |
3.6 kb |
55 °C |
|
42 |
ePDZb (145.3 ng/µl) |
o14sd_005 |
014sd_006 |
Pfu |
ePDZb_Eco_out |
3.4 kb |
55 °C |
|
43 |
CAT-1 mutated |
o14sd_037 |
014sd_038 |
Pfu |
CAT-1_AgeI_out |
5.8 kb |
55 °C |
|
44 |
WPRE RFC 10 |
o14sd_059 |
014sd_060 |
Pfu |
WPRE_NgoMIV_out |
2.6 kb |
55 °C |
|
45 |
P2A RFC 10 (107.3 ng/µl) |
o14sd_027 |
014sd_028 |
Pfu |
P2A_NgoMIV_out |
2.2 kb |
55 °C |
Pfu protocol: basically just a normal PCR program: 1x 95 °C 30 seconds
15x: 95 °C 30 seconds
55 °C 1 minute
68 °C 2 minutes/kb
1x: 68 °C 10 minutes
Afterwards, PCRs weren’t digested by DpnI, but 5 µl were taken for transformation on either Ampicillin or Chloramphenicol agar plates.
2014.09.14
3 transformations worked (PCR 40, 42, 44). Colonies of PCR 42 were small, so they were left an additional day in the incubator.
2014.09.15
|
PCR # |
Template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
46 |
GAL4 |
o14sd_011 |
014sd_012 |
Q5 |
GAL4 RFC 25 |
0.5 kb |
55 °C |
PCR was made in 50 µl and in triplicates.
2014.09.16
Triplicates of PCR 46 were digested in CutSmart by EcoRI-HF/PstI-HF for ligation into pSB1C3. Ligation of PCR 46 was made as last time reported.
ONC were made from PCR 40 (2x), 42 (6x), 44 (2x).
2014.09.17
DNA extraction of ONCs.
|
Template |
concentration [ng/µl] |
Template |
concentration [ng/µl] |
|
ePDZb_Eco_out I |
135.6 |
ePDZb_Eco_out VI |
313.9 |
|
ePDZb_Eco_out II |
185.2 |
SEAP_NgoMIV_out I |
151.0 |
|
ePDZb_Eco_out III |
62.0 |
SEAP_NgoMIV_out II |
200.0 |
|
ePDZb_Eco_out IV |
117.0 |
WPRE_NgoMIV_out I |
84.0 |
|
ePDZb_Eco_out V |
63.9 |
WPRE_NgoMIV_out II |
115.8 |
Test digests
|
Template |
enzyme |
buffer |
fragment size |
|
WPRE_NgoMIV_out |
XhoI/NgoMIV |
CutSmart |
???? |
|
SEAP_NgoMIV_out |
NgoMIV_BamHI-HF |
CutSmart |
Mutated: 3 kb, 1.7 kb Not mutated: 3 kb, 0,9 kb, 0.8 kb |
|
ePDZb |
EcoRI-HF/XbaI/NheI |
CutSmart |
mutated: 2.4 kb, 1.4 kb not mutated: 2.4 kb, 0.9 kb. 0.5 kb |
Ligation of GAL 4 RFC 25 didn’t work.
2014.09.18
Every test digest failed.
Ligation of GAL4 RFC 25 repeated, but with Quick ligation buffer.
|
Plasmid |
backbone |
GAL4 RFC 25 |
|
length (bp) |
2070 |
441 |
|
concentration [ng/µl] |
64.1 |
87.9 |
|
volume [µl] |
0.78 |
0.36 |
|
Water |
|
1.20 |
|
plus 2.6 µl 1x T4 buffer and 0.2 µl T4 ligase (400,000 U/ml) |
||
We used for a 2nd approach instead of molare ratio of 1:3 just thrice volume of insert (0.73 µl pSB1C3 + 1.47 µl GAL 4 RFC 25).
Ligations were incubated at room temperature for 15 minutes. 5 µl were taken for transformation and afterwards plates were incubated at 37 °C.
|
PCR # |
Template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
47 |
pKM292 re-transformation |
o14sd_011 |
014sd_012 |
Q5 |
GAL 4 RFC 25 |
0.6 kb |
55 °C |
|
48 |
p14zl_003 |
o14sd_029 |
014sd_030 |
Q5 |
Puromycin acetyl transferase |
3.6 kb |
55 °C |
|
49 |
WPRE RFC 10 seq. |
o14sd_059 |
014sd_060 |
Q5 |
WPRE RFC 10 with RFC 25 overhangs |
0.6 kb |
55 °C |
Each PCR was made in 50 µl and triplicates.
Gel electrophoresis showed fine results. Lanes were cut out and ligations into pSB1C3 were made at 21.09.
2014.09.21
|
PCR # |
Template |
primer 1 |
primer 2 |
polymerase |
product |
size |
annealing temperature |
|
50 |
CAT-1 mutated |
o14sd_041 |
014sd_042 |
Pfu |
CAT-1 RFC 10 with RFC 25 overhangs |
1.9 kb |
55 °C |
|
51 |
SEAP_PstI_out |
o14sd_051 |
014sd_052 |
Pfu |
SEAP RFC 10 with RFC 25 overhangs |
1.6 kb |
50 °C |
Each PCR was made in 50 µl and triplicates.
ONC were made from ligations.
2014.09.22
No PCR product visible. PCR 50 & 51 will be repeated with Phusion instead of Pfu with a common annealing temperature of 50 °C.
|
PCR # |
Template |
primer 1 |
primer 2 |
polymerase |
Product |
size |
annealing temperature |
|
54 |
PCR 1.1 II |
o14sd_005 |
014sd_006 |
Phusion |
ePDZb_EcoRI_out |
1.9 kb |
55 °C |
PCR was digested by DpnI and afterwards 3 µl were taken for transformation. The bacteria culture was streaked out onto a Ampicilin plate.
DNA extraction of ONC
|
Template |
concentration [ng/µl] |
Template |
concentration [ng/µl] |
|
WPRE RFC 10 (PCR 49) I |
105.0 |
Puromycin acetyltransferase RFC 25 I |
131.4 |
|
WPRE RFC 10 (PCR 49) II |
146.0 |
Puromycin acetyltransferase RFC 25 II |
161.2 |
|
WPRE RFC 10 (PCR 49) III |
131.0 |
SEAP_NgoMIV_out II Puromycin acetyltransferase RFC 25 III |
70.0 |
|
WPRE RFC 10 (PCR 49) IV |
267.0 |
Puromycin acetyltransferase RFC 25 IV |
124.0 |
|
WPRE RFC 10 (PCR 49) V |
140.0 |
Puromycin acetyltransferase RFC 25 V |
230.0 |
|
GAL 4 RFC 25 I |
156.1 |
||
|
GAL 4 RFC 25 II |
247.8 |
||
|
GAL 4 RFC 25 III |
134.4 |
||
|
GAL 4 RFC 25 IV |
135.7 |
||
|
GAL 4 RFC 25 V |
130.4 |
Test digsts of ONC
|
Template |
enzyme |
buffer |
fragment size |
|
WPRE RFC 25 |
NcoI-HF |
CutSmart |
1.5 kb, 1.2 kb |
|
GAL 4 RFC 25 |
XhoI |
CutSmart |
1.2 kb, 0.9 kb, 0.3 kb |
|
Puromycin acetyltransferase |
XhoI |
CutSmart |
1.8 kb, 0.9 kb |
All digests showed lanes with the expected sizes.
2014.09.23
Transformation with PCR 54 worked. Three colonies were picked for ONC.
Gel electrophoresis of PCR 50 & PCR 51

test digest of PCR 50 & 51.
Gel electrophoresis of PCR 50 & PCR 51
img src="https://static.igem.org/mediawiki/2014/2/2a/Freiburg2014_2014-09-23_testverdauPCR50_PCR51_neu.jpg" alt="Description of Image">test digest of PCR 50 & 51.
2014.09.24
DNA extraction of ONC
|
Template |
concentration [ng/µl] |
|
ePDZb_Eco_out I |
49.6 |
|
ePDZb_Eco_out II |
67.7 |
|
ePDZb_Eco_out III |
52.7 |
Test digest
|
Template |
enzyme |
buffer |
fragment size |
|
ePDZb_EcoRI_out |
EcoRI-HF/ClaI/HindHIII-HF |
CutSmart |
Mutated: 2.3 kb, 1.5 kb Not mutated: 2.3 kb, 0.9 kb, 0.6 kb |
Result: all 3 samples showed expected DNA lanes. Colony 2 will be sequenced.
Colony 1 of WPRE RFC 25, GAL 4 RFC 25 and Puromycin acetyltransferase RFC 25 will be sequenced.
2014.09.25
All biobricks showed expected DNA sequence and ePDZb lost the EcoRI site.
|
PCR # |
Template |
primer 1 |
primer 2 |
Polymerase |
Product |
size |
annealing temperature |
|
55 |
ePDZb_EcoRI_out |
o14sd_001 |
014sd_002 |
Phusion |
ePDZb_AgeI_out |
3.8 kb |
55 °C |
|
56 |
SEAP_PstI_out (367.6 ng/µl) |
o14sd_045 |
014sd_046 |
Phusion |
SEAP_NgoMIV_end |
5.8 kb |
50 °C |
|
57 |
SEAP_PstI_out (367.6 ng/µl) |
o14sd_049 |
014sd_050 |
Phusion |
SEAP RFC 10 |
1.6 kb |
50 °C |
|
58 |
WPRE RFC 10 (25.09.) |
o14sd_055 |
014sd_056 |
Phusion |
WPRE RFC 25 |
1.6 kb |
50.5 °C |
Gel of PCR 57 showed a strong lane. The lane was cut out and prepared for digest/ligation. Digest was done with EcoRI-HF/Pst-HF in CutSmart.
Ligation of PCR 50 and PCR 57
|
Plasmid |
backbone |
CAT-1 RFC 25 overhangs |
SEAP RFC 10 |
|
length (bp) |
2070 |
1945 |
1577 |
|
concentration [ng/µl] |
64.1 |
68.7 |
80.4 |
|
volume [µl] |
0.38 |
1.84 |
1.71 |
|
Water |
|
1.20 |
|
|
plus 2.6 µl 1x T4 buffer and 0.2 µl T4 ligase (400,000 U/ml) |
|||
Additional approach: just use a triple volume of insert each (0.6 µl backbone, 1.8 µl insert).
2014.09.26
Transformation of PCR 58 didn’t work but PCR 55 & 56 showed colonies on plates. 5 colonies were picked for ONC.
Ligations were partly successful. There for both SEAP RFC 10 and CAT-1 with RFC 25 overhangs colonies. 5 of them were picked for ONC each.
2014.09.27
DNA extraction from OVC
|
Template |
concentration [ng/µl] |
Template |
concentration [ng/µl] |
|
PCR 56 I |
160.4 |
PCR 55 I |
81.1 |
|
PCR 56 II |
122.9 |
PCR 55 II |
71.4 |
|
PCR 56 III |
109.9 |
PCR 55 III |
46.8 |
|
PCR 56 IV |
105.2 |
PCR 55 IV |
43.1 |
|
PCR 56 V |
199.3 |
PCR 55 V |
81.0 |
|
SEAP RFC 10 I |
182.2 |
PCR 50 I |
130.2 |
|
SEAP RFC 10 II |
100.9 |
PCR 50 II |
120.2 |
|
SEAP RFC 10 III |
122.7 |
PCR 50 III |
17.2 |
|
SEAP RFC 10 IV |
239.0 |
PCR 50 IV |
128.2 |
|
SEAP RF 10 V |
201.6 |
PCR 50 V |
170.3 |
Test digests
|
Template |
enzyme |
buffer |
fragment size |
|
PCR 50 |
EcoRV-HF/KpnI-HF |
CutSmart |
2.2 kb, 1.3 kb |
|
PCR 55 |
AgeI-HF/XbaI/NheI |
CutSmart |
mutated: 2.4 kb, 1.4 kb not mutated: 2.4 kb, 0.9 kb, 0.5 kb |
|
PCR 56 |
NgoMIV/BamHI-HF |
CutSmart |
mutated: 3.0 kb, 1.7 kb not mutated: 3.0 kb, 1 kb, 0.7 kb |
|
PCR 57 |
NcoI-HF |
CutSmart |
1.8 kb, 1 kb, 0.7 kb |
Gel electrophoresis

test digest of PCR 50, 55-57.
PCR 50, 56 and 57 failed. PCR 55 looked like the plasmid had been cut once. Sequence analysis showed that XbaI site was overlapped with a methylation site, thus restriction site was blocked and couldn’t recognized anymore. Therefore PCR 55 I would be sequenced.
2014.09.28
|
PCR # |
Template |
primer 1 |
primer 2 |
Polymerase |
Product |
size |
annealing temperature |
|
59 |
WPRE RFC 10 -NgoMIV |
o14sd_055 |
014sd_056 |
Phusion |
WPRE RFC 25 |
0.6 kb |
57 °C |
|
60 |
CAT-1 PstI out |
o14sd_039 |
014sd_040 |
Phusion |
CAT-1 RFC 10 |
1.9 kb |
56 °C |
PCR 59 was done in 50 µl approaches and in triplicates and was separated in a 2 % gel.
Gel electrophoresis

test digest of WPRE RFC 25 ligation into pSB1C3.
Results looked fine. The PCR product was cut out with EcoRI-HF/PstI-HF and used for ligation into pSB1C3.

test digest of PCR 60.
Results looked fine. The PCR product was cut out with EcoRI-HF/PstI-HF and used for ligation into pSB1C3.
Gel extraction
|
Template |
concentration [ng/µl] |
|
WPRE RFC 25 |
76.2 |
|
CAT-1 RFC 10 |
289.6 |
|
SEAP RFC 10 |
like PCR 57 |
2014.09.30
Sequencing results confirmed that ePDZb is AgeI-free.
|
PCR # |
Template |
primer 1 |
primer 2 |
Polymerase |
Product |
size |
annealing temperature |
|
61 |
PCR 55 I |
o14sd_007 |
014sd_008 |
Phusion |
ePDZb RFC 10 |
0.6 kb |
56.5°C |
|
62 |
SEAP PstI out |
o14sd_045 |
014sd_046 |
Phusion |
SEAP_C2784G_NgoMIV |
5.4 kb |
56 °C |
PCR Product was purified by gel extraction and cut with EcoRI-HF/PstI-HF and used for ligation into pSB1C3.
PCR 62 was digested by DpnI and transformed on agar plate containing Ampicillin.
ligation
|
|
WPRE RFC 25 (09/29) |
CAT-1 RFC 10 (09/29) |
SEAP RFC 10 (09/25) |
|
pSB1C3 |
1.46 µl |
1 µl |
1 µl |
|
insert |
0.74 µl |
0.4 µl |
0.4 µl |
|
water |
/ |
0.8 |
/ |
|
plus 2.6 µl Quick ligation buffer, 0.2 µl T4 ligase (400,000 U/ml) |
|||
2014.10.01
All transformed plates had colonies.
DNA extraction
|
Template |
concentration [ng/µl] |
|
WPRE RFC 25 I |
130.0 |
|
WPRE RFC 25 II |
109.0 |
|
WPRE RFC 25 III |
115.0 |
|
WPRE RFC 25 IV |
140.0 |
|
PCR 62 I |
448.0 |
|
PCR 62 II |
458.0 |
|
PCR 62 III |
490.0 |
|
PCR 62 IV |
413.0 |
|
Template |
concentration [ng/µl] |
Template |
concentration [ng/µl] |
|
SEAP RFC 10 I |
216.9 |
CAT-1 RFC 10 I |
265.9 |
|
SEAP RFC 10 II |
192.9 |
CAT-1 RFC 10 II |
396.3 |
|
SEAP RFC 10 III |
193.3 |
CAT-1 RFC 10 III |
223.2 |
|
SEAP RFC 10 IV |
176.5 |
CAT-1 RFC 10 IV |
272.2 |
|
SEAP RFC 10 V |
167.5 |
CAT-1 RFC 10 V |
209.0 |
|
SEAP RFC 10 VI |
193.4 |
CAT-1 RFC 10 VI |
188.6 |
|
ePDZb RFC 10 I |
125.1 |
||
|
ePDZb RFC 10 II |
144.4 |
||
|
ePDZb RFC 10 III |
117.5 |
||
|
ePDZb RFC 10 IV |
125.0 | ||
|
ePDZb RFC 10 V |
118.8 |
||
|
ePDZb RFC 10 VI |
109.5 |
Test digests
|
Template |
enzyme |
buffer |
fragment size |
|
PCR 59 |
EcoRV-HF/NgoMIV |
CutSmart |
1.7 kb, 0.9 kb |
|
PCR 60 |
EcoRV-HF/KpnI-HF |
CutSmart |
2.2 kb, 1.4 kb, 0.4 kb |
|
PCR 61 |
XhoI |
CutSmart |
1.8 kb, 0.9 kb |
|
PCR 62 |
BamHI-HF/NgoMIV |
CutSmart |
2.3 kb, 1 kb, 0.3 kb |
Gel electrophoresis

test digest of PCR 62 and ligation of WPRE RFC 25 into pSB1C3.
All results looked fine. It seemed that SEAP contained a NgoMIV site less and the WPRE digests proved that WPRE RFC 25 is in pSB1C3.

test digest of following ligations: ePDZb RFC 10, SEAP RFC 10, CAT-1 RFC 10.
Results looked fine. Every sample showed expected lane size.
Colony one of each plasmid was sent to sequencing.
Standardization - October
2014.10.02
All four ligations contain expected DNA sequence.