 "
"
Team:SUSTC-Shenzhen/Notebook/Parts building
From 2014.igem.org
Parts building
design and building
Contents |
By Rifei Chen
For the linear backbone were so few and the backbone of BBa_J04450 was pSB1C3, we decide to cut it by XbaI and PstI restriction enzyme and recycle the about 2000bp vector as the backbone for biobricks. Of course, one advantage to make it as backbone is that we can recognize whether the gene we designed have inserted. Because if not, the bacterium will express red chromoprotein which one we unnecessary to pick up.
Week 4, Aug
2014/08/22
Transfect the backbone of biobricks
- Add 10μl ddwater to the plate, wait 10min, and absorb all the liquid into a PCR tube.
- Took a tube of supercompetent cells from -80℃ fridge, Immediately place the tube on ice, allow cell to thaw on ice, 10 min.
- Check the cell to see if they have thawed, gently flick the cells 1-2 times to evenly resuspend the cells.
- Mark the label with BBa_J04450.
- Add 2μl plasmid to the tube, shaking slightly.
- Incubate on ice for 30min.
- Heat shock, 42℃, 90s.
- Keep on ice immediately, 2 min.
- Add 200μl LB medium to each tube, shake with 37°C, 200rpm, 45min.
- Centrifuge, 4000rmp, 3min at room template.
- Discard most of the medium and reserve 50μl, resuspend by tips.
- Flam the SS-Spreader; put aside to cool; coated plates.
- Incubate the plates at 37°C, 16 hours.
2014/08/23
Shake the bacterium in tubes
- Add 3ml LB broth with ampicillin antibiotics into a tube.
- Flam the tweezers to red; put aside to cool.
- Pick up a tip which was sterilized; pick monoclone and throw the tip with monoclone into the tube directly.
- Shake with 37°C, 200rpm, and overnight.
Week 5, Aug
2014/08/24
Plasmid purification
- Pour 1.5ml of broth into a centrifuge tube, 11000 rpm, 2min.
- Repeat step1 again.
- Using TIANprep Mini Plasmid Kit, remove the medium completely, add 150μl P1 buffer, resuspend by tips, votex.
- Add 150μl P2 buffer, up and down gently 6~8 times; till it turn clear.
- Add 350μl P5, immediately invert up and down 10~12 times; till it clear and appear flocks. 11000rpm, 5 min.
- Transfer the supernatant into absorption column CP3, do not carry the precipitate. 11000rpm, 1 min. Discard the liquid waste.
- Add 600μl PW buffer; wait 3min; 11000rpm, 1min. Discard the liquid waste.
- Repeat step7 again.
- Centrifuge again at 11000 rpm, 2 min. Discard the liquid waste.
- Put the absorption column CP3 into a new centrifuge tube, open the cover and wait alcohol to spread completely.
- Add 70μl TB buffer into the absorption column center; wait 2min, 11000rpm, 2min.
- Re-suck up TB buffer which were centrifuged and add into the absorption column center again; wait 2min, 11000rpm, 2min.
- Measure the concentration of plasmid by Nanodrop2000. Mark in the tube.
2014/08/28
Design the biobricks
Week 2, Sep
2014/09/07
Design the primers
2014/09/11
Place an order of primers and completely synthesis
- Primers show:
SV40 promoter For: T TCTAGA G CGAACTGTGGAATGTGTG
SV40 promoter Rev: A GGATCC AT GAAGAC GACGAAAATGGATATACAAG
SV40 PolyA For: G AGATCT AT GAAGAC CCAACTTGTTTATTGCAGCTTA
SV40 PolyA Rev: CTGCAG CGGCCGC T ACTAGT ATCCATGCCGAGAGTGATGAA
pTRE-3G promoter For: T TCTAGA G TTTAAACTTTACTCCCTATC
pTRE-3G promoter Rev: A GGATCC AT GAAGAC GATTTACGAGGGTAGGAAGTG
TetOn For: T TCTAGA TGGGATCAAGACTGGACAAGA
TetOn Rev: CTGCAG CGGCCGC T ACTAGT A CCGAAGCCCAACCTTTCATA
CBh promoter For: T TCTAGA G TCTAGAGGTACCCGTTACA
CBh promoter Rev: A GGATCC AT GAAGAC GTCCAACCTGAAAAAAAGTGA
PB3 For: G AGATCT AT GAAGAC TGCTAGACTATAACAAGAA
PB3 Rev: CTGCAG CGGCCGC T ACTAGT A CGAAATTAACCCTCACTAA
PB5 For: T TCTAGA G ATTAGAAACTATTATTTAAC
PB5 Rev: A GGATCC AT GAAGAC GACACACATTCCACAGTTCG
- Completely synthesis show:
HBV1:
5'GATCAAGAAGACCTCACCAGGACCCCTGCTCGTGTTACAGGGTTTGGGTCTTCGATCTC 3'
HBV2:
5'GATCAAGAAGACCTCACCTCCGCAGTATGGATCGGCAGGTTTGGGTCTTCGATCTC 3'
HIV:
5'GATCAAGAAGACCTCACCTCTAGCAGTGGCGCCCGAACAGGGTTTGGGTCTTCGATCGA
5*UAS:
5'CGGAATTCGCGGCCGCTTCTAGAGCGGAGTACTGTCCTCCGAGCGGAGTACTGTCCTCCG
ACTCGAGCGGAGTACTGTCCTCCGATCGGAGTACTGTCCTCCGCGATTTCCGGAGTACTGTC
CTCCGTACTAGTAGCGGCCGCTGCAGAACCAATGCATTGG 3'
7*UAS:
5'CGGAATTCGCGGCCGCTTCTAGAGCGGAGTACTGTCCTCCGCGGAGTACTGTCCTCCGGA
ATTGCGATAGGTACCGAGTTACTAGACGGAGTACTGTCCTCCGAGCGGAGTACTGTCCTCCG
ACTCGAGCGGAGTACTGTCCTCCGATCGGAGTACTGTCCTCCGCGATTTCCGGAGTACTGTC
CTCCGTACTAGTAGCGGCCGCTGCAGAACCAATGCATTGG 3'
PolyA:
5'GAAGATCTTCGAAGACCTAGAGCTCGCTGATCAGCCTCGACTGTGCCTTCTAGTTGCCAG
CCATCTGTTGTTTGCCCCTCCCCCGTGCCTTCCTTGACCCTGGAAGGTGCCACTCCCACTGT
CCTTTCCTAATAAAATGAGGAAATTGCATCGCATTGTCTGAGTAGGTGTCATTCTATTCTGG
GGGGTGGGGTGGGGCAGGACAGCAAGGGGGAGGATTGGGAAGAGAATAGCAGGCATGCTG
GGGATACTAGTAGCGGCCGCTGCAGAACCAATGCATTGG 3'
NLS+NLS:
5'TCTGGAATTCGCGGCCGCTTCTAGAGCCAGATGGCCCCAAAGAAGAAGCGGAAGGTCGGT
ATCCACGGAGTCCCAGCAGCCACGTCTTCATGGATCCTATGAAGACTGAAAAGGCCGGCGGC
CACGAAAAAGGCCGGCCAGGCAAAAAAGAAAAAGTACTAGTAGCGGCCGCTGCAGTCCGGCA
AAAAAGGGC 3'
Week 3, Sep
2014/09/18
PCR TetOn, PB5, PB3, SV40/CBh/pTRE-3G promoter, PolyA1, 2*UAS
For TetOn and CBh were more than 700bp, we decided to use Q5® High-Fidelity DNA Polymerases, but others by OneTaq® 2X Master Mix. 2*UAS without template because the forward primer and reverse primer coμld combine each other and extend.
The PCR systems set by protocol show below, we made 50μl reaction.
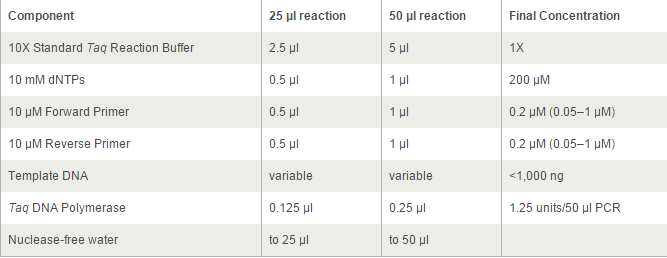
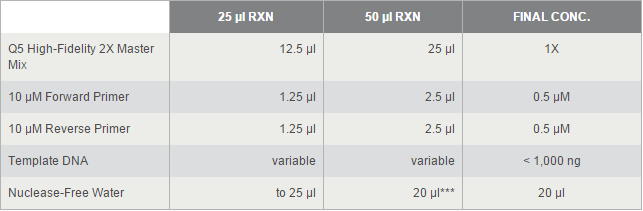
Thermocycling Conditions for a Routine PCR:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 90 |
| 30cycles | 98 | 30 |
| 48,52,55,58,60,63 | 30 | |
| 68 | 30 | |
| Final extension | 68 | 300 |
| Hold | 4 | ∞ |
Make a 1% Agarose gel:
| Component | Volume |
|---|---|
| 1×TAE buffer | 50ml |
| Agarose | 0.50g |
| Gene Green | 0.5ul |
Gel electrophoresis result:


PB3 and PB5 were both failed.
Purified others by TIANgel Midi Purification Kit. Use Nanodrop2000 to get each sample’s concentration. Digest TetOn, 2*UAS and backbone by XbaI and PstI overnight; CBh/SV40/pTRE-3G promoter by XbaI and BamHI; PolyA1 by BglII and PstI; purified.
The digest systems:
| Enzyme(μl) | Buffer(μl) | DNA(ng) | ddH2O(μl) | Total volume(μl) |
|---|---|---|---|---|
| 0.5+0.5 | 2.5 | 500 | up to 25 | 25 |
2014/09/19
Ligation and transformation
The ligation systems (vector + TetOn, vector + 2*UAS, vector + CBh/SV40/pTRE-3G promoter + PolyA1) :
| T4 ligase(μl) | T4 DNA ligation buffer(μl) | Vector and insert mole ratio | ddH2O(μl) | Total volume(μl) |
|---|---|---|---|---|
| 1 | 2 | 1:5/1:5:5 | up to 20 | 20 |
16℃ for 35min, inactive at 65℃ for 10min.
Transfect with DH5α
Re-PCR PB3 and PB5, still without any bands.
2014/09/20
Shake the bacterium in tubes
Pick up backbone + TetOn, backbone + 2*UAS monoclone and shake overnight.
There were no bacteria grown of backbone + CBh/SV40/pTRE-3G promoter + PolyA1.
Analyzed and find the primers of CBh/SV40/pTRE-3G promoter and PolyA1 were all out of protect bases. Re-design and order new primers.
Week 4, Sep
2014/09/21
Plasmid purification and digest to check length, sequencing
Purified plasmids by TIANprep Mini Plasmid Kit. Digest by XbaI and PstI for two hours. Make a 1% Agarose gel. The results show right length
2014/09/22
Send TetOn and 2*UAS to sequencing
2014/09/24
Re-PCR CBh/SV40/pTRE-3G promoter, PolyA1, PB3, PB5 by new primers and digest
The same PCR systems and reaction conditions like 2014/09/18, but CBh promoter and PB5 failed for wrong length. SV40/ pTRE-3G promoter have many bands for the primers’ low specificity. Still purified CBh and PB5.
Purified by TIANgel Midi Purification Kit. Use Nanodrop2000 to get each sample’s concentration.
Digest CBh/SV40/pTRE-3G promoter and PB5 by XbaI and BamHI; PolyA1 and PB3 by BglII and PstI overnight.
The digest systems:
| Enzyme(μl) | Buffer(μl) | DNA(ng) | ddH2O(μl) | Total volume(μl) |
|---|---|---|---|---|
| 1+1 | 2.5 | 1000 | up to 25 | 25 |
2014/09/25
Re-ligase CBh/SV40/pTRE-3G promoter + PolyA1, PB5+PB3 and transformation
The ligation systems (vector + CBh/SV40/pTRE-3G promoter + PolyA1 and vector + PB5 + PB3) :
| T4 ligase(μl) | T4 DNA ligation buffer(μl) | Vector and insert mole ratio | ddH2O(μl) | Total volume(μl) |
|---|---|---|---|---|
| 1 | 2 | 1:5:5 | up to 20 | 20 |
16℃ for 35min, inactive at 65℃ for 10min.
Transfect with DH5α
Sequencing show TenOn was right but 2*UAS lost one SpeI site.Analyzed and find due to the wrong direction of primer. Re-design and order new primers.
2014/09/26
Shake the bacterium in tubes
Pick up monoclone and shake overnight. PB5 + PB3 failed for without any monoclone.
2014/09/27
Purified CBh/SV40/pTRE-3G promoter + PolyA1 and digest, check by gel electrophoresis
Purified plasmids by TIANprep Mini Plasmid Kit. Digest by XbaI and PstI for two hours. Make a 1% Agarose gel.
Gel electrophoresis result:

All results get wrong.
2014/09/30
Digest UAS, HIV, HBV, PolyA2, NLS-NLS
Get completely synthesis sequence plasmid and digest UAS, HIV, HBV, PolyA2, NLS-NLS overnight.
The digest systems of 5*UAS/7*UAS:
| Enzyme XbaI+PstI(μl) | Buffer 2.1(μl) | DNA(ng) | ddH2O(μl) | Total volume(μl) |
|---|---|---|---|---|
| 0.5+0.5 | 2.0 | 400 | 7 | 20 |
The digest systems of HIV/HBV1/HBV2/PolyA2/NLS-NLS:
| Enzyme XbaI+PstI(μl) | Buffer 2.1(μl) | DNA(ng) | ddH2O(μl) | Total volume(μl) |
|---|---|---|---|---|
| 0.5+0.5 | 2.0 | 225 | 7 | 20 |
Touchdown PCR for CBh promoter, PolyA1, PB3 and PB5
The PCR system show:
| Taq polymerase(μl) | 0.5 |
|---|---|
| Buffer(μl) | 5 |
| dNTP(μl) | 1 |
| Primer For(μl) | 0.5 |
| Primer Rev(μl) | 0.5 |
| Template(μl) | 1 |
| ddH2O(μl) | 41.5 |
| Total(μl) | 50 |
Thermocycling Conditions for a Routine PCR:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 95 | 180 |
| 15 touchdown cycles each cycle down 1℃ of annealing temperature | 95 | 15 |
| 55 | 30 | |
| 72 | 40 | |
| 35cycles | 95 | 15 |
| 45 | 30 | |
| 68 | 40 | |
| Final extension | 68 | 300 |
| Hold | 4 | ∞ |
Gel electrophoresis result:

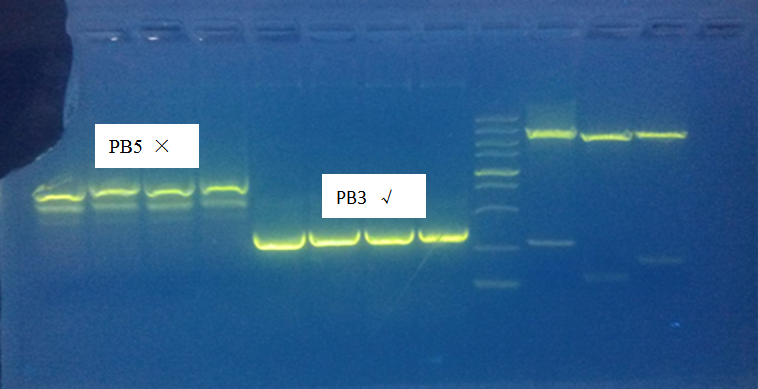
Only PolyA1 and PB3 were PCR successfully, CBh and PB5 were wrong length. But still purified and digest overnight.
PCR 2*UAS by new primers. Check, purify and digest overnight.
Week 1, Oct
2014/10/01
Ligase UAS, HIV, HBV, NLS-NLS, PB3+PB5 to backbone
Purified the digest plasmids and PCR products by TIANgel Midi Purification Kit. Use Nanodrop2000 to get each sample’s concentration.
Ligase UAS/gRNA for HIV/gRNA for HBV/NLS-NLS with backbone by mole ratio 7:1 at 16℃ for 35min, and then inactive at 60℃ for 10min.
Ligase PB3+PB5, CBh/SV40/pTRE-3G promoter+PolyA1, CBh/SV40/pTRE-3G promoter+PolyA2 with backbone by mole ratio 7:10:1 at 16℃ for 35min, and then inactive at 60℃ for 10min.
Transfect ligation plasmid but increase the incubation time to 2 hours.
2014/10/02
Pick up the monoclony to do colonyPCR and shake
CBh promoter + PolyA do not grow the right bacterium for all grown bacterium show red chromoprotein. So decide give it up.
Pick up 5 monoclonies each sample of SV40/pTRE-3G promoter+PolyA1, SV40/pTRE-3G promoter+PolyA2 and a colonyPCR to check whether the length were rigth.
Gel electrophoresis result:

Picture shows SV40/pTRE-3G promoter+PolyA1 were all right length but SV40/pTRE-3G promoter+PolyA2 were all wrong.Shake three sample each
Pick up a monoclony each sample of 2*UAS, HIV, HBV
Gel electrophoresis result:
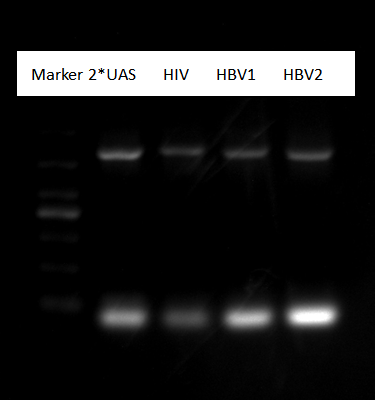
Picture shows all sample were right.Shake them in tubes.
2014/10/03
Purified SV40/pTRE-3G promoter + PolyA1 and digest, check by gel electrophoresis
Purified plasmids by TIANprep Mini Plasmid Kit. Digest by XbaI+PstI, XbaI+BbssI for two hours. Make a 1% Agarose gel and wait 1h to be solid.
Gel electrophoresis result:
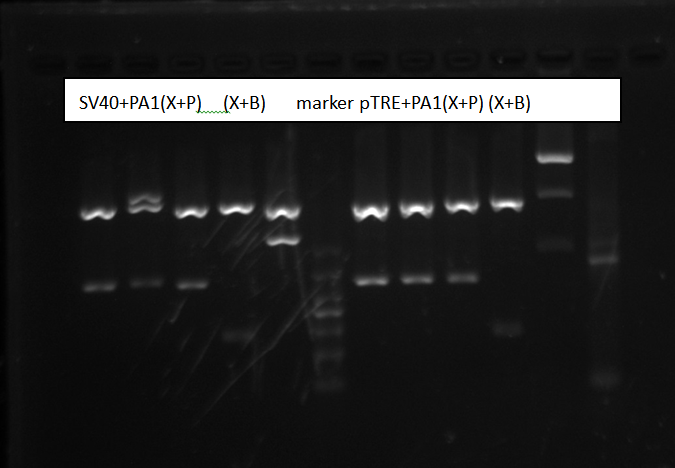 Picture shows all sample were right. We both chose the sample3 to send to sequencing.
Picture shows all sample were right. We both chose the sample3 to send to sequencing.
Of course send UAS, gRNA and NLS to sequencing.
PCR the target sequence of HIV, HBV1, HBV2.And the next three days do the same thing like above, purify, digest, ligase, sequencing.
Week 2, Oct
2014/10/08
The last sample send to sequencing
2014/10/09
All plasmids make dry and send to the HQ of iGEM
2014/10/10
PCR eGFP for SV40 and pTRE-3G
The PCR system show:
| Ex Taq polymerase(μl) | 0.25 |
|---|---|
| Buffer(μl) | 5 |
| dNTP(μl) | 4 |
| Mg2+(μl) | 4 |
| Primer For(μl) | 0.5 |
| Primer Rev(μl) | 0.5 |
| Template(μl) | 1 |
| ddH2O(μl) | 34.75 |
| Total(μl) | 50 |
Thermocycling Conditions for a Routine PCR:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 180 |
| 35cycles | 98 | 15 |
| 52,57,61 | 30 | |
| 68 | 60 | |
| Final extension | 68 | 300 |
| Hold | 4 | ∞ |
Took 1μl each sample to make a gel electrophoresis, observed all the aim bands PCR successfully. By TIANgel Midi Purification Kit, we get the purified sequence.
2014/10/11
Digest and ligase SV40/pTRE-3G promoter + eGFP
Digest SV40, pTRE-3G, eGFP for SV40 and pTRE-3G by BbsI restriction enzymes for 3hours. By TIANgel Midi Purification Kit, get the vector of SV40 and pTRE-3G. Use Nanodrop2000 to get the concentration. Ligase the vector and insert by mole ratio 1:5 at 16℃ for 35min, and then inactive at 60℃ for 10min.
Transfect ligation plasmids but increase the incubation time to 2hours.
2014/10/12
Colony PCR of SV40/pTRE-3G promoter + eGFP
Pick up 8 monoclone from SV40 promoter + eGFP plate, 8 monoclone from pTRE-3G promoter + eGFP, and 1 monoclone as active control group. Each monoclone dissolve in 10μl ddH2O respectively, and suck 1.5μl as the template.
Gel electrophoresis result:
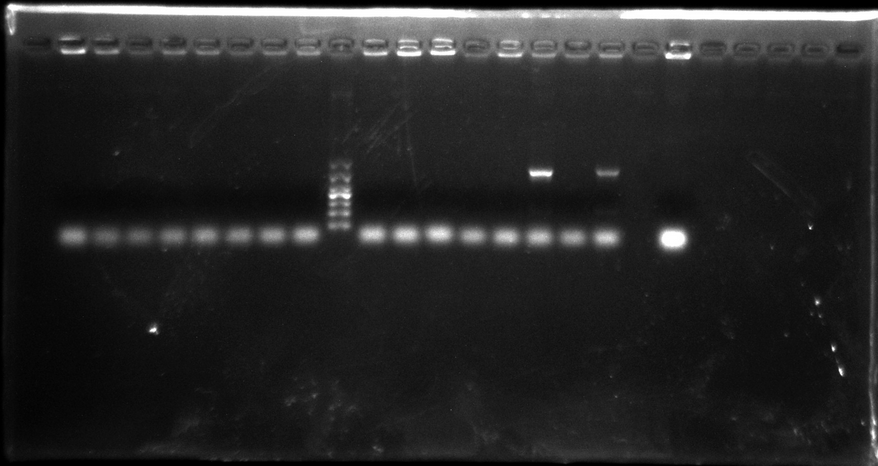
There were only two samples PCR successfully, and not right length. So we think that may be the PCR system got wrong. We decide to do that again but change system.
Re-Colony PCR of SV40/pTRE-3G promoter + eGFP
The same thinking as above, but do with another kind of taq polymerase.
Pick up 6 monoclone from SV40 promoter + eGFP plate, 6 monoclone from pTRE-3G promoter + eGFP, and 1 monoclone as active control group. Each monoclone dissolve in 10μl ddH2O respectively, and suck 1.5μl as the template.
Gel electrophoresis result:

All the PCR results were wrong for the wrong length. I doubt the restriction enzymes did not cut the vector or insert.
For the limited time, we decide not to do that again.