 "
"
Team:UChicago/Project
From 2014.igem.org

Our Project
Introduction
Directed evolution is a fundamental technique in bioengineering organisms to express proteins of novel function and to mass produce industrially relevant biomolecules. Generally, directed evolution simulates an algorithmic process in which the entire sequence space is searched for an optimal genotype by increasing the natural mutation rate to artificially speed the process of selective evolution. Current methods of in vivo directed evolution typically rely on physical or chemical mutagens to accomplish stochastic genomic or plasmid mutagenesis. However, the extremely high and uncontrolled mutation rate often accumulates deleterious mutations nonspecific to the process of interest. This can result in a toxic effect to the organism, leading to suboptimal evolved levels of production.
A novel directed evolution system, termed feedback-regulated evolution of phenotype (FREP) (citation http://www.ncbi.nlm.nih.gov/pubmed/24131951), incorporates a dynamic mutation rate to overcome the existing problems of directed evolution by mimicking the plasticity of the mutation rate in natural evolution. This is achieved through dynamic control of a mutator element that is negatively regulated by the desired end product. In this feedback scheme, as more of the desired biomolecule is produced, the rate of mutation decreases and eventually approaches zero, allowing evolution and maintenance of a high level of production while minimizing the accumulation of toxic, nonspecific mutations.
Additionally, many directed evolution systems that incorporate mutator genes often rely on the strongest known mutator, MutD. However, the use of MutD alone in these systems is problematic because like all other individual mutator genes, it may only catalyze certain types of base pair substitutions reflective of its “mutational bias”, limiting the search of sequence space. Diversifying the mutator genes to eliminate mutational bias should increase the efficiency by which the sequence space can be searched. Ultimately, the addition of multiple mutators should lead to higher overall levels of evolved production. Despite a very few number of past studies to incorporate multiple mutators, these efforts have failed to show an increased efficiency of evolution. They fail to encompass a systematic screen of all mutators identified in literature, which would be required for determination of the ideal combination of mutators. In general, there is a surprising lack of studies on the use of different combinations of mutators in directed evolution.
The 2014 UChicago iGEM team has two main goals. The first goal is to implement and optimize FREP in E. coli by evolving mutants with elevated production of a given biomolecule, specifically evolving tyrosine production as a relatively simple pathway to demonstrate proof of concept. The mutator element will be controlled by the tyrosine-sensitive repressible promoter ParoF, suppressing the mutation rate as the tyrosine production of the cell increases. The mutator gene is linked to a fluorescent reporter gene which allows for convenient screening of desired phenotype, as bacteria with low levels of fluorescence should also have the highest levels of production due to the negative feedback loop. The second goal is to enable a deeper exploration in the search space by combining multiple mutators with different biases to increase the initial mutation rate and speed the accumulation of beneficial mutations. We have identified previously characterized mutator genes that function in a diversity of pathways, including polymerases, proofreading enzymes, methylases and topoisomerases. We will characterize the mutation rates of these mutator genes in their active dominant negative forms individually as well as in various combinations, aiming to determine the ideal combination for directed evolution. We hope to introduce this ideal combination of mutators into the FREP system and demonstrate increased levels of evolved tyrosine production compared to the single mutator. If successful, our project should significantly improve the process of directed evolution of any biomolecule of industrial importance.
Mutator Constructs
In order to work towards our goal of finding the ideal multiple mutator gene combination for FREP, we first researched a variety of genes that when overexpressed, had been previously documented in literature to increase the mutation rate. Because of the manner in which FREP is specifically set up, dynamic control can only be achieved in a gene that increases mutation rate when overexpressed (as opposed to mutators that cause an increase in mutation rate from knockdown or full deletion for instance). Since most of the mutator genes we used had little effect from simple overexpression, we instead relied on mutation dominant negative versions of these genes that are documented to cause high increases in mutation rate when expressed in cells. These mutator genes are involved in a variety of pathways, and include polymerase subunits, DNA repair enzymes, and transposable elements. We also kept in mind the various mutational biases of the various mutators we were interested in. The table below lists all various types of mutators that we researched, as well as the point mutations needed to create the active dominant negative genes when necessary.; bolded mutators with an asterisk (*) are the actual mutators we ended up attempting to PCR from the wild-type E. Coli genome. (citation)
Mutators Studied
| Mutator gene | Mechanism (pathway) | Mutation rate (estimate) If available, relative to wild type? | Bias | Point Mutations (when necessary) | References |
|---|---|---|---|---|---|
| MutD* | mutD (dnaQ) gene product controls the editing capacity of pol III (a proofreading DNA exonuclease) mutD5 mutation leads to a reduction in mismatch repair | 10^3-10^4 errors per base pair per cell per generation Editing efficiency: WT 0.059 mutD 0.012 Mutation rate 10^5 higher than WT | Affects all bases, effect limited by temperature and richness of medium (works better in less rich media) Mostly transversions | MutD51: L73W This looks stronger than the real MutD5, which has a second suppressor mutation (below). We may want to do both though since it’s really not that much more work. MutD52: A164V | [1] [2] [3] [4] |
| MutT | Nucleoside triphosphatase Works by reducing the 8-oxo-dGTP level in the cell through hydrolysis to 8-oxo-dGMP | 10^3-10^4 errors per base pair per cell per generation | AT to CG Transversions mutT excludes A-G mismatches where the adenine resides on the template DNA strand. | None found | Link to various journal articles |
| MutS* | Primarily recognizes base mismatch sites, works with MutL, MutH to repair mismatch sites (MutS-1, at least). Seems to detect changes in adjacent base pairs truncated form of the DNA mismatch repair protein | 2.5 ± 2.0 × 10^–6 (?) | G619D | [1] [2] [3] | |
| MutM | removes 8-oxoG residues from DNA by DNA glycosylase activity | > 10^2 | GC to TA Transversions -combining mutM and MutY seems to increase mutation rate 25-75X compared to either mutator alone (see paper) | None found | [1] |
| emrR* | involved in multidrug resistance, transcription regulator | 2.8 * 10^-4 //// 2.8 * 20^4 | AT->GC preferred transversions + frameshift (tested +1G) | NA (overexpression of wild-type gene sufficient) | [1] |
| Dam* | involved in methylating DNA | 6.4 * 10^-4 /// 6.4 * 10^4 | Transversions | NA (overexpression of wild-type gene sufficient) | [1] |
| bglY-mutant (bglY-galU region) | deletions, recessive, negative supercoiling? | 2 to 10^2 | Unknown | None found | [1] |
| MutL* | Along with MutS and MutH, initiates repair cascade by recognizing base mismatches/indels of up to 4 bp (dam-directed mismatch repair) | 2.75*10^-8 bp/generation 138 X wild type | Unknown | E32K | [1] [2] [3] |
| DinB* | Error-prone DNA polymerase DinB, mutagenesis error-prone polymerase IV | NA | increase in frameshift and base substitution | F13V SImple overexpression is also possibly mutagenic | [1] [2] |
| Umu2D’C* | DNA checkpoint effector, encodes pol V | NA | hypersensitivity to UV light damage in mutants | Y11A Simple overexpression is also possibly mutagenic | [1] |
| MutH* | Mismatch repair,nvolved in the same pathway as MutS and MutH | 10^2-10^3 | GC to AT, AT to GC, Frameshifts | MutH: E56A | [1] [2] |
| IS transposable elements | More old-school transposons, generally exert effect by altering expression by insertion in promoter regions -May also vary the number of IS elements introduced | ~10X compared to wild type | [1] | ||
| Mini transposons | Mini-Mu, Mini D-108, mini-Tn5, mini-Tn7 Could test 1-2 of these. | Variable | Can induce larger-scale chromosome rearrangements | [1] | |
| DnaE* | alpha subunit of DNA polymerase 3 | E612K | [1] | ||
| ParC* | subunit of topIV | Y120H | [1] | ||
| MutY* | G-A mismatch DNA repair | > 10^2 | GC to TA Transversions | V45A | [1] |
Though we ran out of time and were never able to test actual combinations of mutators (we were only able to test individual mutators), it would make more sense to combine mutators with different biases (one could combine a mutator with a G to C bias with one with an A to T bias for example) as opposed to mutators with similar biases, which we would predict to allow greater coverage of the sequence space.
Cloning Strategy
To engineer the mutators into the final FREP system, we divided up the project into several parts. First, the individual mutators were cloned into an IPTG-inducible promoter construct, and mutation rates of individual mutators were quantified in this construct, simultaneously allowing verification of their mutagenic activity. In a separate construct, the ParoF tyrosine-repressible promoter that normally controls the AroF-containing operon was cloned into the plasmid backbone. Finally, a mutated version of the TyrR transcription factor that regulates ParoF was to be created on a third plasmid; the mutations would alter and increase the range of tyrosine concentrations that TyrR was sensitive to.
All initial cloning of the mutators from the wild-type E. Coli genome and site-directed mutagenesis when necessary was done in the IPTG-inducible promoter construct. This design facilitated creating combinations of mutators in the ParoF construct, because we could design universal primers that allowed us to easily PCR out the mutagenized mutators once they had been created in the IPTG-inducible promoter construct, followed by sequential cloning of mutators in the desired order on the ParoF-containing construct.
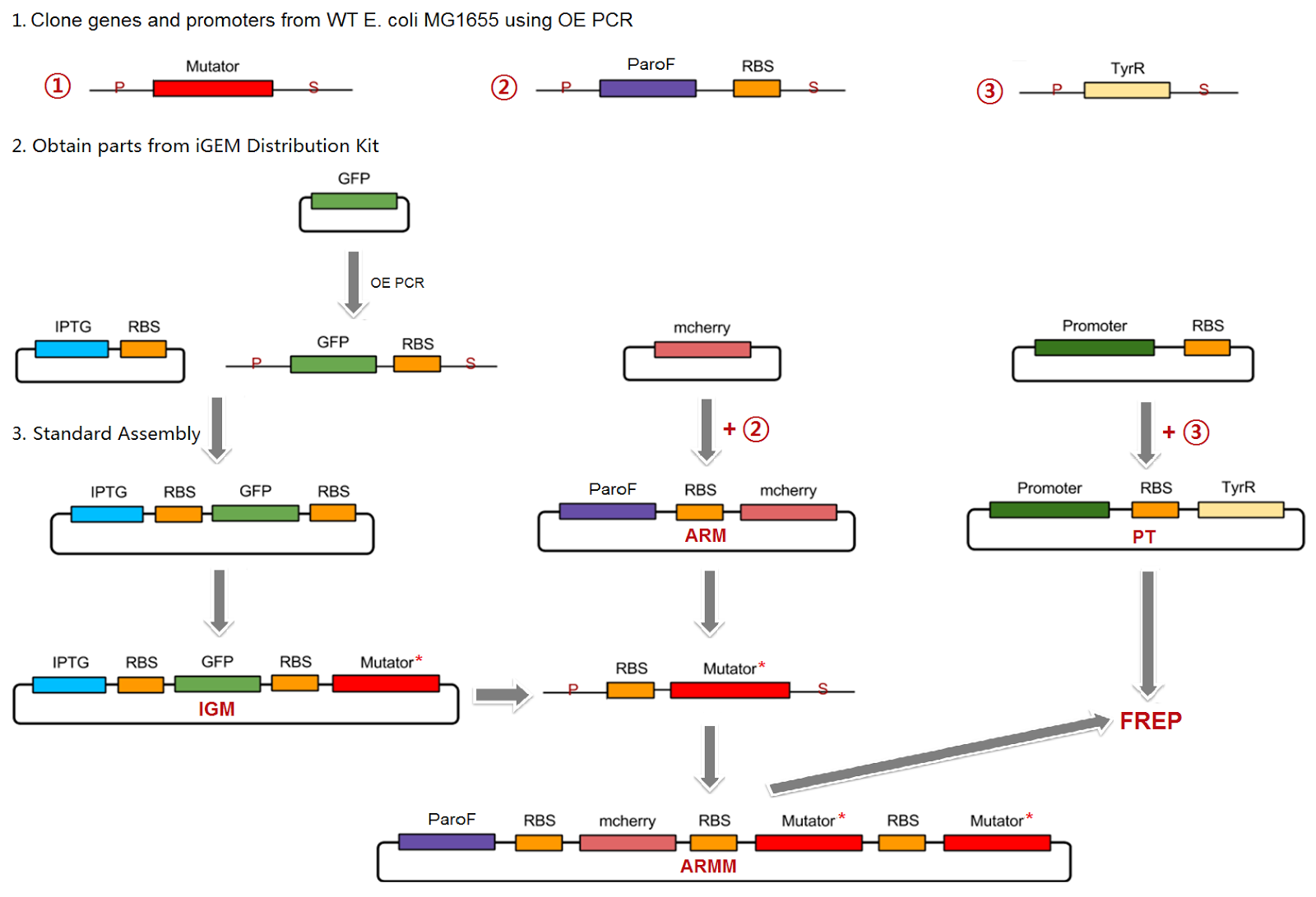
To read about the specific details and functional units of each of the general constructs we used, click here.
To read about the calculations and assays used to quantify mutation rate and verify the repressibility of the ParoF construct, click here.
Results
We were able to isolate the mutator genes EmrR, Dam, mutD, mutL, DnaE, mutS, mutY, and mutH from E. coli genomic DNA and place them into a construct containing GFP under the control of an IPTG-inducible promoter. We then activated mutS,mutY, and mutH into their mutation-causing alleles via Quikchange mutagenesis (wild type Dam and EmrR can raise the mutation rate of E. coli when simply overexpressed)
We were then able to quantify the mutation rate of XL1Blue (Agilent) bacteria containing the IPTG->GFP+EmrR plasmid and compare it to that of wild type Xl1Blue. Using a modified version of the Luria-Delbruck fluctuation analysis experimental setup (quantifing mutation rate through estimating number of mutational events through frequency of rifampicin-resistance) and online FALCOR tool (http://www.keshavsingh.org/protocols/FALCOR.html), we were able to estimate the mutation rate per cell per culture for both strains.
| mact=(mobs*(1-z))/(z(ln(z)) | |||||
|---|---|---|---|---|---|
| rif-resistant colonies per parallel culture | avg#cells per culture | mutations(observed) | mutations (actual) | Mutation rate mutations/cell per culture) | |
| wt | 1,3,1,4,1,4,1,1,1,3,6,4,1,1,0,0 | 1.98E+09 | 1.217 | 5.617 | 2.84E-09 |
| EmrR | 1,2,3,1,1,1,1,0,0,0,0,0,0,0,0,0 | 5.95E+08 | 0.478 | 1.868 | 3.14E-09 |
The FALCOR tool uses a maximum likelihood function to generate an estimate for number of mutational events from the frequency of rif-resisitant colonies in parallel cultures. We had to correct the estimate of mutations m(observed) to m(actual) (using mact=(mobs*(1-z))/(z(ln(z)) with z being fraction of cultures plated) because we only plated 10% of each parallel culture onto the rifampicin selective media. The mutation rate is gained by dividing the actual number of mutations that occurred by the average number of cells per culture. Because the cell densities were different, this data is not wholly reliable as bacteria may have quorum sensing abilities that may impact endogenous mutation rate (http://www.nature.com/ncomms/2014/140429/ncomms4742/full/ncomms4742.html).
We successfully created FREP-ready ARM constructs containing the mutagenizing proteins EmrR, Dam and mutH(E56A) and the reporter mCherry-LVA under the control of the tyrosine-sensitive promoter paroF und although we were not able to implement FREP due to time constraints.