 "
"

9/3/14
A 1mL glycerol stock of bacteria containing pET28YCLY4 of equal part cells and a 60% glycerol stock was prepared from sample 1 of the overnight culture and stored in the -80°C freezer.The two pET28YCLY4 samples were minipreped following the Miniprep Protocol with some exceptions. The overnight culture was centrifuged at 3900rpm for 10 minutes for the first step of preparing the cell lysate. For the second step of eluting the DNA, 50μl of preheated TE buffer was added to the column instead of the stated 75μl.
After the plasmids were isolated, the concentration was measured using the nanodrop. Samples 1 and 2 of pET28CLY4 was measured to be 62.5 and 47.4 ng/μl, respectively. The plasmids were stored at -20°C.
9/4/14
Rosetta cells were transformed with the pET28CLY4 plasmid following the general transformation protocol and plated. One sample was made for each of the pET28CLY4 sampled minipreped as well as a negative control. 50μl of cells, 100μl of SOC media and 2μl of plasmid (or dH2O for the negative control) were used for each sample.9/5/14
The plated cultures were checked and are shown below.Each plate had small and large colonies which is unusual for dissimilar sized colonies to grow. Because colonies of the negative control grew, the transformations were redone with the same protocol.
9/6/14
The plates were checked for colonies. There were a few colonies growing in the negative plate but since the number of colonies was much less than than those of the pET28CLY4 plates, it was attributed to degradation of the antibiotic over time. A colony from a pET28CLY4 plate was restreaked and incubated overnight.


9/15/14
Inoculate 2 sets of minicultures with pET28+_TdT, pSB1C3 and pSB1A3 from glycerol stocks. Grow overnight 37°C with shaking9/16/14
One of the pSC1A3 cultures failed to grow. Follow the miniprep procedure on remaining cultures. Nanodropped and ran 500ng on a gel.
Left to right: Ladder | pSB1C3-A | pSB1C3-B | pSB1A3 | pET28+_TdT-A | pET28+_TdT-B
Set up PCR to amplify TdT out of pET28+_Tdt. Use First truncated forward and regular reverse. Also, set up two aliquots of 20μL reaction.
9/17/14
Ran gel of yesterday's PCR
Left to right: Ladder | neg control | pET28+_TdT-A | pET28+_TdT-A | pET28+_TdT-B | pET28+_TdT-B
Seeing a product in the negative control, as well as the intensity of the bands in the sample wells, we assumption one of the reagents has a DNA fragment or a PCR product. We will repeat the PCR using all new aliquots of reagents and primers.
9/18/14
Ran a gel of yesterday's PCR
Left to right: Ladder | empty | neg control | pET28+_TdT-A | pET28+_TdT-A | pET28+_TdT-B | pET28+_TdT-B
While there is a smear in the negative control, our target fragment was amplified and is the correct size. Additionally, the smear is smaller than the target fragment, so it should be good to gel purify.
After gel purification, a second PCR was ran with the purified fragment, using the new δEcoRI primer and the regular reverse.
9/19/14
Ran a gel of yesterday's PCR
Left to right: Ladder | neg control | pET28+_TdT-A | pET28+_TdT-A | pET28+_TdT-B | pET28+_TdT-B
Bands are very faint, but in the correct location. Gel extract the samples from this gel, and run the remainder of the PCR reaction on a second gel to extract the target band. Nanodrop confirms the yield is low.
9/22/14
Double digest the PCR purified fragments, and the new miniprepped pSB1C3, using XbaI and SpeI. Phosphotase treat the vector before setting up the ligation. Run ligation at room temperature for 30 minutes, then transform into NEB5α.9/23/14
Colonies grew on the plates; negative controls had no growth. Parafilm and put in the refridgerator to see if any red colonies develop.9/24/14
A few colonies turned red, but most of the colonies are white. Pick 5 colonies to miniprep tomorrow.9/25/14
From the TdT CleanAmp-dNTP Comparison Experiment, it was shown that in longer reaction incubation times produced longer oligos. To get a clearer sense of the enzymatic reaction rate, the TdT Enzymatic Reaction Rate Experiment protocol was followed. Because TdT would add 30+ bases to the starting oligo in just 10 minutes, the incubation times were reduced so the products would fit between the oligo ladder on the gel so that less extrapolation would be needed to determine a reaction rate.
TdT was allowed to react for 1-12 minutes. The gel from gel electrophoresis of the products seem to suggest a linear relationship between the incubation time and bases added. A enzymatic reaction rate was calculated from the experiment's results.
10/17/14
Rosetta cells transformed with pET28b+_TdT were IPTG induced to express TdT. The enzyme was extracted and purified. The Expressed TdT Experiment protocol was followed.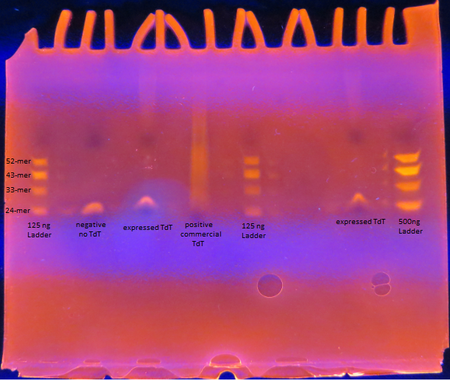
As seen from the gel, the TdT expressed from the Rosetta cells with our plasmid works as intended; base pairs are appended to oligos by the enzyme. There is a difference between the activity though between our TdT and commercially obtained TdT however. The majority of the oligos have only a few base pairs added by our expressed TdT while the lane of the commercial TdT is a smear of oligos, suggesting the commercial TdT has a greater reaction rate. But it can also be attributed to the fact that the 0.5 U/μL concentration of our expressed TdT is a overestimation. The TdT concentration of the reaction with our expressed TdT is less than that of the commercial TdT.
There is also a large gap between the oligos with only a few bases added and those with 30+ bases added. This suggests that our expressed TdT has a higher processivity than that of commercially obtained TdT. This might be due to subtle sequence differences such as the His tag on our TdT sequence.



