 "
"
Team:SUSTC-Shenzhen/Project/gRNA-mCherry-UAS Carried Plasmids Design and Construction
From 2014.igem.org
gRNA-mCherry-UAS Carried Plasmids Design and Construction
In this part, we have totally constructed 16 plasmids, to transport the gRNA sequences into human cells.
Abstract
There are three types of gRNA-carried related plasmids. Each type carries out a different function.
The first four plasmids which carry the gRNA for EGFP aim to check if the CRISPR-Cas9 system we create in human cell can work well, which might be the foundation for the further experiments. The following three plasmids carried no gRNA sequences but two BbsI sites are capable for the insertions of any wanted gRNA sequences. As for the last nine plasmids, actually they come from the type two plasmids, but have been inserted with gRNA for HIV/HBV. Thus, they can be used to target for HIV/HBV gene sequence that has been integrated in human chromosome specially.
However, although each type with different missions, they still have plenty of components in common. You can see, nearly all of them carry at least one UAS sequences, since UAS sequences are necessary for binding with the non-viral DNA delivery vehicle. And also, mCherry-3*NLS is another DNA sequence which appears in all of the 16 plasmids. This is because this part plays an important role in efficiency test, both for gRNA transportation and cut-off ability of CRISPR-Cas9.
Following are three profiles of plasmids, served as examples for each type:
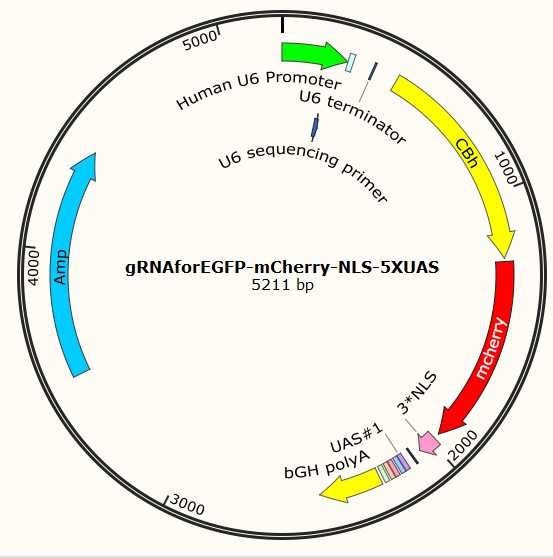
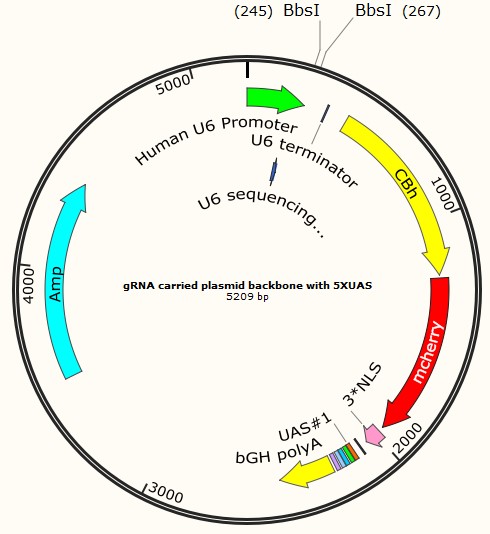
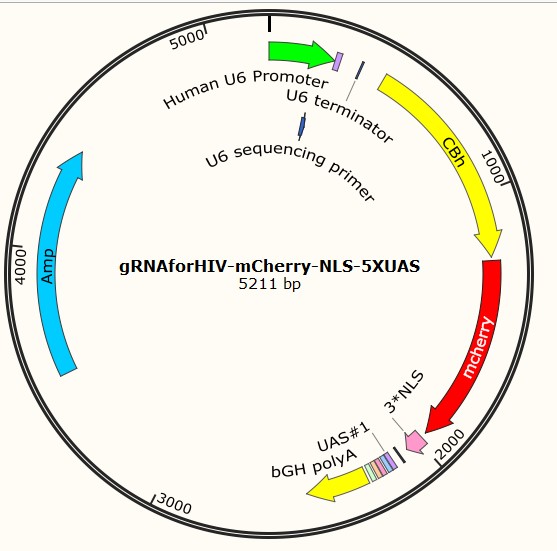
Part I Plasmid construction Design: Why do we construct them?
Why do we design plasmids that only carry gRNA sequence without Cas9?
One main thought of our project is to achieve the transferring of Cas9 system and gRNA sequences into human body separately at different period of time.
As you know, there are two parts in our CRISPER system, one is the Cas9 proteins, and the other is the gRNA. To begin with, Cas9 system will be stably transfected into human cells firstly, remaining inactivated and just waiting for orders. While the transportation of gRNA will not be performed until the diseases have developed, and types of retrovirus that cause these diseases are confirmed. Then, since the type of the pathogenic retrovirus has been known at this moment, we can design a gRNA that can specifically recognize the very type of retrovirus, and transfer it into human body. Finally these gRNA will combine with activated Cas9 proteins, guiding them to the genome sequence of retrovirus, at last destroying it.
By this way, we can achieve a more specific introduction of gRNA into human cells, specifically targeting for the very type of retrovirus that caused the disease. The separated transportations of the two parts may also partially overcome the difficulties of radical cure, always met in usual treatment, due to the high variability of retrovirus.
That’s why we constructed these plasmids, and each of which are used to carry gRNA sequence into human cells. Among them, four have already carried gRNA sequences, thus can be transferred directly [gRNAforEGFP-pX330-mCherry-NLS-0XUAS, gRNAforEGFP-pX330-mCherry-NLS-2XUAS, gRNAforEGFP-pX330-mCherry-NLS-5XUAS, gRNAforEGFP-pX330-mCherry-NLS-7XUAS]. While the other three are only designed with BbsI site, preparing for the specific gRNA sequence insert [BbsISiteForgRNA-pX330-mCherry-NLS-2XUAS,BbsISiteForgRNA-pX330-mCherry-NLS-5XUAS, BbsISiteForgRNA-pX330-mCherry-NLS-7XUAS].
Why dose each of our plasmids carries at least one UAS sequence?
We choose a non-viral DNA delivery system which is based on modified A-B toxin with a yeast GAL4 transcription factor, to deliver our plasmids carried with gRNA into human cells.
Yeast transcriptional activator GAL4 protein can specifically recognize plasmids containing UAS sequences and bind with them. Thus, by replacing A subunit of A-B toxin with GAL4, we will get a chimeric fusion protein that can transfer plasmids with UAS into host cells specifically.
Therefore, it is necessary for our plasmids to carry at least one UAS sequence, so that they can be recognized, and at the same time combine with the DNA delivery shuttle, and finally be transported into human cells.
Why do we construct plasmids with different number of UAS sequences?
When GAL4 binds with UAS carried plasmids, their binding affinity may differ among plasmids that carry different number of UAS sequences. To test the binding efficiency of GAL4 part in our DNA delivery vehicle, we totally constructed four plasmids, each of which carried 0X, 2X, 5X, 7X UAS sequences respectively. By comparing their binding results with the GAL4, we can screen out the most efficient one with the optimum UAS number for binding. Therefore, in the following construction of specific gRNA carried plasmids, we can use this number as a standard, to achieve the most effective transportation.
Why dose each of our plasmids still carries a mCherry-3*NLS sequence?
Although it seems that the mCherry-3*NLS sequence in our plasmid has little contribute to our CRISPR-Cas9 working system, this part of DNA sequence dose make sense, when we plans to test the working efficiency of the whole system.
Before using our non-viral DNA delivery system, we need to test the delivery efficiency of this chimeric protein system at first. So, we add the mCherry-3*NLS gene sequence just behind a human U6 promoter in our plasmids. If our plasmids have been successfully transported into human body cells, the 3*NLS sequences will lead these plasmids into the nuclei. After a series of processes like transcription, translation and modifications, mCherry red fluorescence protein(RFP) will finally be expressed and excitated a bright red fluorescence under the fluorescence microscope. In this way, we can visualize how many plasmids on earth are transferred into nuclei, just by detecting and counting the number of red-fluorescence-emitting cells.
What’s more, with this RFP expression mechanism we are able to roughly estimate the cut-off efficiency of the CRISPR-Cas9 system, by using the ratio between amount of green fluorescence reduced and quantity of red fluorescence generated. [Please see Cell Culture part for more details about CRISPR-Cas9 system efficiency test]
Why do we also construct plasmids with no gRNA sequences but two BbsI restriction enzyme sites?
Since we want to achieve a specific introduction of gRNA that can specifically target for the very type of retrovirus, it is reasonable to come up with the idea about constructing a plasmid which is convenient for various sequences of gRNA insertions. This plasmid will act as a backbone that carries the appropriate restriction enzyme sites for gRNA insertion and all the other necessary parts (except gRNA sequence) in the former plasmids.
For this reason, we replace the gRNA sequences in our former plasmids with a DNA fragment that carries two BbsI sites, to achieve the insertions of any gRNA sequences we wanted. With this gRNA carried plasmid backbone, we can introduce any gRNA sequences exactly for the specific retrovirus at any time, partially overcoming the difficulties of radical cure, which are always met in usual treatment, due to the high variability of retrovirus.
Part II Method: How do we construct them?
A. Construction of gRNA-for-EGFP carried plasmids
Preparation of pX330 plasmid backbone:
All of the first seven plasmids we constructed are based on the plasmid pX330 (8000bp, HongKong) We use EcoRI, AgeI & EcoRV to do enzyme digestion of plasmid pX330, removing the entire Cas9 sequence and collect the remaining 4000bp DNA fragment as ligation vector for the following experiments.
Point-mutation PCR for mCherry-3*NLS sequence from plasmid pBX-084- PB5-HS4-TRE-mCherrynuc-2A-BlaloxN-GpA-HS4-PB3 (from Wei Huang’s lab):
We acquire our mCherry-3*NLS DNA sequence (800bp) by PCR from plasmid pBX-084.
With primers A, we achieve:
a. Get the mCherry-3*NLS sequence, also with EcoRI and AgeI enzyme sites added on each end.
b. Do Point-mutation PCR at the same time, successfully eliminating the BbsI site on it.
Primers A:
Forward (5’-3’): TCGTTTAGTGAACCGTCAG
Reverse (5’-3’): cgaggaattc ctatta TTCCTCTGCCCTCACTAG
With primers B, we achieve:
In addition to a and b,
c. Get mCherry-3*NLS-2XUAS sequence, by adding 2XUAS sequence behind the mCherry-3*NLS.
Primers B:
Forward (5’-3’): TCGTTTAGTGAACCGTCAG
Reverse (5’-3’): cgaggaattc cggaggacagtcctccg cggaggacagtcctccg ctatta TTCCTCTGCCCTCACTAG
cgaggaattc cggaggacagtcctccg cggagtactgtcctccg ctatta TTCCTCTGCCCTCACTAG
Point-mutation PCR for 5XUAS sequence from plasmid PB5-PolyA-5XUAS (HongKong):
We get the 5XUAS sequence by PCR from plasmid PB5-PolyA-5XUAS (HongKong):
With primers C, we achieve:
a. Get the 5XUAS sequence, also with MfeI enzyme sites added on each end.
b. Do Point-mutation PCR at the same time, eliminating the XbaI, AgeI & EcoRI enzyme sites on it.
Remark: We also succeeded in mutating out EcoRI site on this DNA fragment by doing 5XUAS Fill-in. [Please see notebook for more details about 5XUAS Fill-in]
Primers C:
Forward (5’-3’): ttaa caattg CGAGTTTCTAGACGGAGTA
Reverse (5’-3’): Taat caattg CGGTCTTAGATCGCAGAT
Construction of gRNA carried plasmids by Restriction enzyme digestion & Ligation:
a. gRNAforEGFP-pX330-mCherry-NLS-0XUAS:
Do enzyme digestion (with EcoRI and AgeI) & ligation for pX330 vector & mCherry-3*NLS sequence. Thus, we get the gRNA carried plasmid with 0XUAS sequence, gRNAforEGFP-pX330-mCherry-NLS-0XUAS.
b. gRNAforEGFP-pX330-mCherry-NLS-2XUAS
Do enzyme digestion (with EcoRI & AgeI) & ligation for pX330 vector & mCherry-3*NLS-2XUAS sequence.
Thus, we get the gRNA carried plasmid with 2XUAS sequence, gRNAforEGFP-pX330-mCherry-NLS-0XUAS.
c. gRNAforEGFP-pX330-mCherry-NLS-5XUAS
We acquire this plasmid by insert the 5XUAS sequence into the plasmid gRNAforEGFP-pX330-mCherry-NLS-0XUAS that we have already constructed.
Do enzyme digestion (with EcoRI & MfeI) & ligation for gRNAforEGFP-pX330-mCherry-NLS-0XUAS & 5XUAS sequence.
d. gRNAforEGFP-pX330-mCherry-NLS-7XUAS
We acquire this plasmid by insert the 5XUAS sequence into the plasmid gRNAforEGFP-pX330-mCherry-NLS-2XUAS that we have already constructed.
Do enzyme digestion (with EcoRI & MfeI) & ligation for gRNAforEGFP-pX330-mCherry-NLS-2XUAS & 5XUAS sequence.
B. Construction of gRNA carried plasmid backbone
Remove the gRNA sequences from the above four gRNA carried plasmids.
Do enzyme digestion for the above four gRNA carried plasmids with PstI & XbaI, removing the gRNA sequences and collect the remaining DNA backbone as ligation vector for the following experiments.
Get the DNA fragment that carries two BbsI sites by enzyme digestion & Ligation:
Do enzyme digestion (with PstI & XbaI) & liagtion for the plasmid pX330 from Prof. Zhang Feng’s lab & the vector collected in the last step.
Thus, we get the gRNA carried plasmid backbones:BbsISiteForgRNA-pX330-mCherry-NLS-2XUAS, BbsISiteForgRNA-pX330-mCherry-NLS-5XUAS, BbsISiteForgRNA-pX330-mCherry-NLS-7XUAS
C. Construction of gRNAforHIV/HBV carried plasmids
gRNAforHIV-pX330-mCherry-NLS-2XUAS
gRNAforHBV1-pX330-mCherry-NLS-5XUAS
gRNAforHBV2-pX330-mCherry-NLS-7XUAS
We acquire them just by inserting the corresponding gRNA for HIV/HBV sequences into the above three gRNA carried plasmid backbones, respectively.
Remark
The results of efficiency test for gRNA transportation and CRISPR-Cas9 cut-off ability can be found in A-B toxin Part & Cell Culture Part.