 "
"
Team:Heidelberg/pages/PCR 2.0
From 2014.igem.org
Contents |
Introduction
Motivation
The invention of the polymerase chain reaction in 1983 by Kary Mulliy revolutionized the modern world by enabling amplification of DNA in an exponential manner. Further improvements including the use of thermo-stable DNA-polymerases made the method even more efficient, allowing its widespread use in nearly every field of modern diagnostics and research. However a major part of information is lost when using conventional PCR due to missing transfer of DNA modifications. Those covalently attached functional groups are present all kingdoms of life and act as prominent regulators of gene expression, adding an additional level of complexity.
We propose the PCR 2.0 as an easy and efficient way to amplify DNA templates in an exponential manner while maintaining their specific methylation pattern. The pivotal element of this PCR reaction is the use of a heat stable DNA methyltransferase (DNMT). Although comparable enzymes exist in thermophile organisms [1] until now no suitable protein could be found or synthesized that withstands the harsh conditions of a PCR.
Even though the detection and mapping of methylation patterns has become feasible in a high-throughput manner by using bisulfite sequencing and array techniques [2], further functional analysis is still limited due to the small amount of primary material. Our approach therefore aims to provide unlimited amounts of any methylated DNA sample of interest, withouth having to know its actual structure or the need of expensive methods and de-novo synthesis – just by running a PCR.
Hence, the main goal of our project has been the generation of a heat stable Dnmt that can be produced in a large scale and shows efficient and specific methylation of DNA. Our approach applies protein circularization by using self-splicing protein sequences, socalled inteins from our toolbox as well as our linker design software. Since the restriction of conformational changes through intramolecular bonds has been known to increase the stability of proteins [3] [4] and joining of C and N terminus were reported to increases in thermostability of smaller peptides [5] we started the circularization of the so far biggest protein…
Epigenetics - there is more than just A, T, C and G…
In mammals and other vertebrates the methylation of cytosine nucleotides that are followed by guanines (CpG) is a prominent key regulator of transcription, embryonic development, X chromosome inactivation and many other cellular functions. [6] [7]. Approximately 1% of the genetic code in human somatic cells is consisting of methylated cytosines, amounting to 70-80% of all present CpG Dinucleotides [8]. Due to this great prevalence and the heredity of this modification, the 5-methylcytosine (5mC) is also known as the “fifth base” of eukaryotic genomes [9]. DNA methylation is preferentially occurring at intergenic regions and repetitive sequences, where it is known to silence gene expression [10] [11]. As against that, GC-rich promoter regions are usually characterized by a low methylation status, allowing transcription of the associated genes [12]. Disregulations of Cytosine methylation have been reported to play a crucial role in the development of numerous diseases, including cancer, imprinting diseases and repeat-instability diseases including morbus huntington [13].
A closer look at DNA Methyltransferases
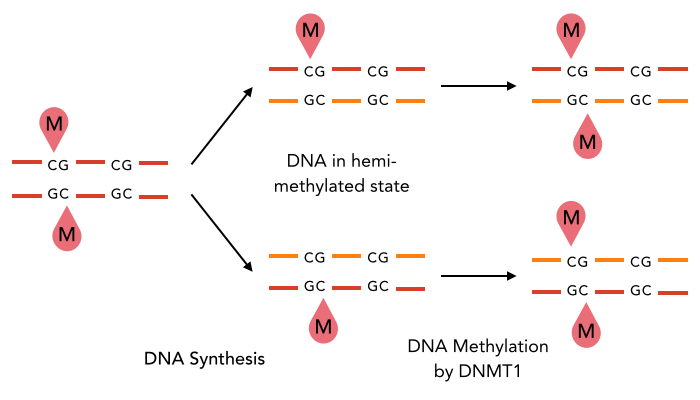
The enzyme DNMT1 is of particular interest for methylation maintenance PCR, as it allows exact duplication of the original methylation pattern to every new DNA copy.
The family of enzymes called DNA methyltransferases (DNMTs) is responsible for the establishment and maintenance of most cell-type specific DNA methylation patterns. Whereas the two enzymes DNMT3a and DNMT3b contribute to de novo methylation of DNA during development, DNMT1 is responsible to preserve existing methylation patterns throughout cell divisions. Therefore DNMT1 exploits the principle of semiconservative replication, using the parental strand as a template to create an exact copy of methylation patterns. Subsequent to recognition of a socalled hemi-methylated CpG site, the enzymatic machinery transferres a methyl group from the methyl donor S-Adeosyl-methionine (SAM) to the C5 position of the complementary cytosine residue. [14]

Structural overview of full length mDNMT1 and truncated mDNMT1(650–1602) (A) Color-coded domain devision and numbering of mDNMT1 sequence. (B) Ribbon presentation of full-length DNMT1 with the CXXC,BAH1, BAH2,and methyltransferase domain colored in light blue, yellow, orange and red, respectively. (C) Crystal structure of truncated DNMT1, missing the CXXC domain. Distant between terminal ends of the proteins are closer than at the full length version.
For our experiments we used the smallest truncated version of DNMT1 that has been reported to efficiently mediate methylation maintenance. This truncated version derives from Mus musculus and comprises amino acids 731-1602 (mDNMT1(731-1602) [15] kindly provided by Dr. Bashtrykov after approval from Prof. Patel). mDNMT1(731-1602) contains the C-terminal catalytic methyltransferasedomain as well as the aligning bromo-adjacent homology (BAH) domains, which prevent the binding of unmethylated DNA in the catalytic core. The enzyme is missing the CXXC domain, which has a high affinity to CpG hemi-methylated dinucleotides, but was shown to be dispensable for protein function [16]. The truncated version has similar methylation activity and specificity as the wild type, as shown by our own experiments and by Bashtrykov and colleagues. In contrast to the full-length DNMT1 that can only be expressed in insect cells using a baculovirus based system, the mDNMT1(731-1602) has the great advantage of being efficiently expressed in E.coli. Since the N and C terminal ends of DNMT1 (731-1602) are closer together than in the full-lengh DNMT1, cirularisation might cause less deformation and have a lower impact on the overall activity, making this truncated mDNMT1 an ideal target for our purposes.
Circularization - The missing link
However, even the truncated DNMT1 is a very large protein of 100kDa and consists of 872 amino acids. To our knowledge, a protein this size has never been circularized before. According to the crystal structure, the distance between the termini of the truncated DNMT1 is 48 Å. We suspected that circularization of the protein by direct fusion of both termini might cause deformation of the protein structure. Therefore we generated a linker amino acid sequence, adapted to the structure of DNMT1: it needed to be long enough to bridge the gap of 48 Å and should bypass the catalytic core located prominently in the middle of the enzyme structure. Since we were not able to estimate the impact of the linker introduction, we focused on two different kinds of linkers: a rather rigid linker that has been calculated and optimized during the establishment of our linker and a flexible peptide connection consisting mostly of Glycine and Serine amino acids. To perform circularization we have used the split NpuDnaE Intein since it is known as the golden standard of protein splicing and has been efficiently tested by us through circularization of GFP, Lysosyme and Xylanase. Moreover, we tried to implement protein circularization by using Sortase A, which is recognizing and cleaving a carboxyterminal sorting signal followed by a transpeptidation reaction that can be exploited for protein circularization. This method has been reported to be very efficient, especially when used in vitro [4].
Material and methods
Constructs
We designed constructs in order to characterize efficiency of sortase as well as intein mediated circularization of DNMT1. Both approaches are based on the comparison of the circularized protein with a corresponding linear counterpart at different temperatures and incubation times.
For circularization of DNMT1 with inteins we fused the obtained truncated version of DNMT1 (731-1602) [15] with an appropriate linker and split NpuDnaE domains at either site of the construct. For efficient purification of the protein, all constructs contained a hexa-histidine tag. Moreover, we have cloned the ubiquitin-like Smt3 protein from Saccharomyces cerevisiae in front of the DNMT1-Intein complex. This attachment is used to increase the yield when expressing large mammalian proteins like DNMT1 in E.coli [17]. The constructs have been designed in a way that Smt3 is no longer included in the final circular protein, and therefore cannot interfere with its function. All cloning steps were carried out using our new RFC(LINK) standard procedure.
The mDNMT1 (731-1602) constructs that have been designed to allow Sortase A mediated cirularization are flanked by the Sortase A recognition sequence LPETGG on the C–terminus as well as N-terminal glycines that that can be exposed through previous TEV treatment. Since the transpeptidation via sortase requires an additional in vitro reaction, subsequent purification is necessary. To facilitate this step and enrich the circular product in the sample, a hexa-histidine tag is present in the initial protein that is lost during circularization. Hence, an additional affinity chromatography via his trap can be used to separate the histidine tag containing sortase and not circularized educt from the circular flow-through. All cloning steps were carried out using the Sortase standard that you can find here (LINK).
The following linkers that had been optimized along with the linker software were used:
| Definition | Sequence |
|---|---|
| Rigid linker | GGAEAAAKEAAAKVNLTAAEAAAKEAAAKEAAAKEAAAKEAAAKAVNLTAAEAAAKAHHHHHHSGRGT |
| Flexible linker | CWEGGGSGGGSGGGSGGGSGGGSGGGSGGGSGGGSGGGSGGGSHHHHHHSGRGT |
Expression and purification
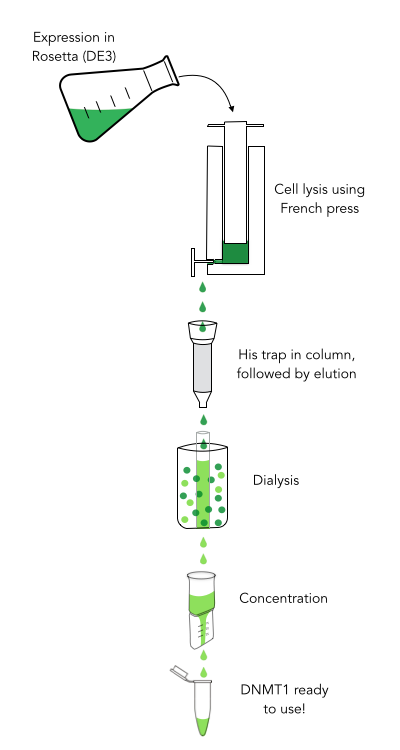
Purification of recombinantly expressed mDNMT1 is a challenging task including multiple steps
In comparison to the full length murine DNMT1, our modified mDNMT1(731-1602) can be expressed in E.coli in a large scale without having to establish a Baculovirus based expression system in insect cells. First experiments were conducted to determine the optimal conditions for protein induction and expression. We tested several concentrations of IPTG as well as different temperatures to maximize the yield of mDNMT1. Still, even though expression of the constructs can be simplified by using E.coli , purification of the recombinant protein remains a challenging task requiring multiple different steps that had to be established.
In a first step, we had to optimize the process of cell lysis, to be able to extract a maximum amount of active protein from our cultures. Therefore we compared the two commonly used methods sonication and disruption via French press. Subsequently, the high sample volume was reduced by ultracentrifugation. The protein of interest containing a hexa-histidie tag was further enriched via immobilized metal ion affinity chromatography (IMAC) using a His trap and eluted with imidazol. In a next step, low molecular weight solutes such as salts and the imidazol that would interfere with the protein function were removed from the sample by dialysis. Finally, the purified protein was concentrated by centrifugation using filters with an appropriate mass cutoff.
Methylation assay
To measure the efficiency of mDNMT1 maintenance methylation, we used an assay that is based on two methylation-sensitive restriction enzymes Sau3AI and HpaII. It relies on the inhibition of restriction enzymes when methyl groups are attached to the cleavage site. We used a 40-mer DNA template with one hemi-methylated and one unmethylated CpG site which are located within the Sau3AI and HpaII restriction site, respectively (adapted from Bashtrykov et colleagues [16].

Sequence of the 40-mer DNA template. CpG dinucleotides are shaded in gray, Sau3AI and HpaII recognition sites are depicted in red. The first CpG dinucleotide is methylated on the reverse strand.
The restriction enzyme Sau3AI is capable of cleaving the template despite of the hemimethylated state of the first CpG nucleotide. Similarly, HpaII can attack its unmethylated native site, leading to fragmentation of the template as long as no further methylation occurs. Accordingly, the methylation activity of circular and linear mDNMT1 can be measured by quantifying the cleavage efficiency of the restriction enzymes after incubation with the protein.
Impairment of Sau3AI cleavage indicates maintenance activity of DNMT1, whereas a decrease in HpaII activity would result from de novo methylation. Therefore the assay is not only detecting activity of the enzyme but also includes its specificity for hemimethylated DNA over unmethylated DNA. To characterize mDNMT1 heat stability as well as kinetics, we perfomed methylation reactions after different heat treatments and looked at the amount of methylation that could be detected due to increasing incubation times.
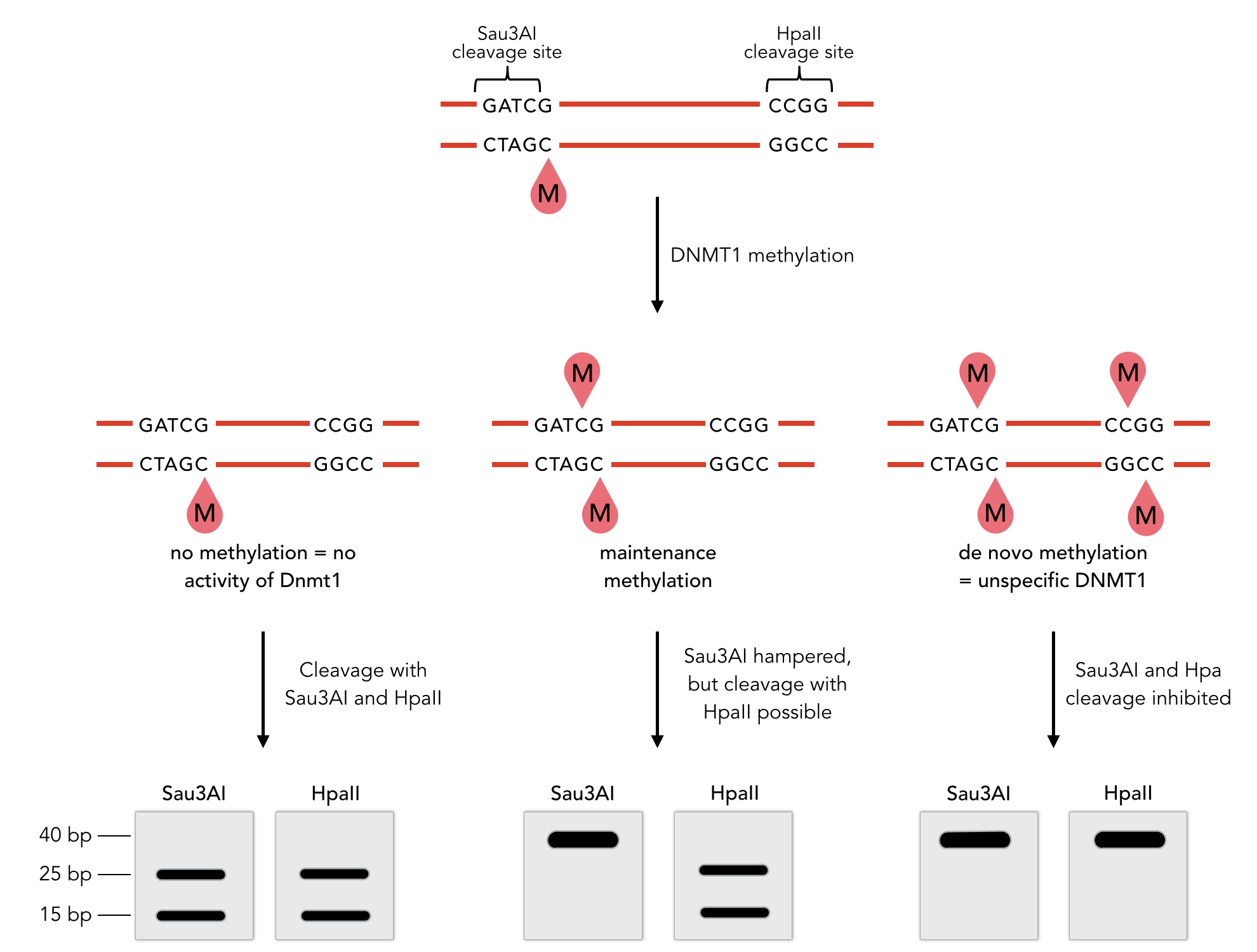
Principle of mDNMT1 methylation assay using a 40mer substrate with Sau3AI and HpaII cleavage sites that are blocked upon further CpG methylation. The lack of methylation activity leads to fragmentation of the DNA template with either of the two restriction enzymes. Whereas specific methylation of hemimethylated CpGs hamperes only cleavage by Sau3AI, unspecific methylation activity of mDNMT1 can be detected when HpaII cleavage is blocked as well.
Assay analysis
For evaluation cleaved DNA was made visible by gel electrophoresis and intensities were measured using ImageJ.
Results

hemi me = negative control, homo me = positive control
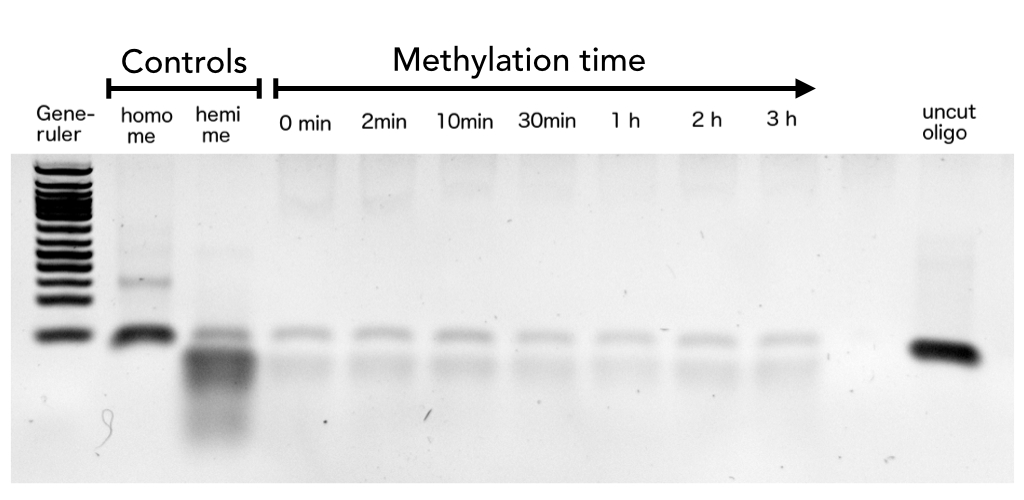
The picture shows HpaII cleavage of the unmethylated restriction site.

Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 55°C and 60°C.
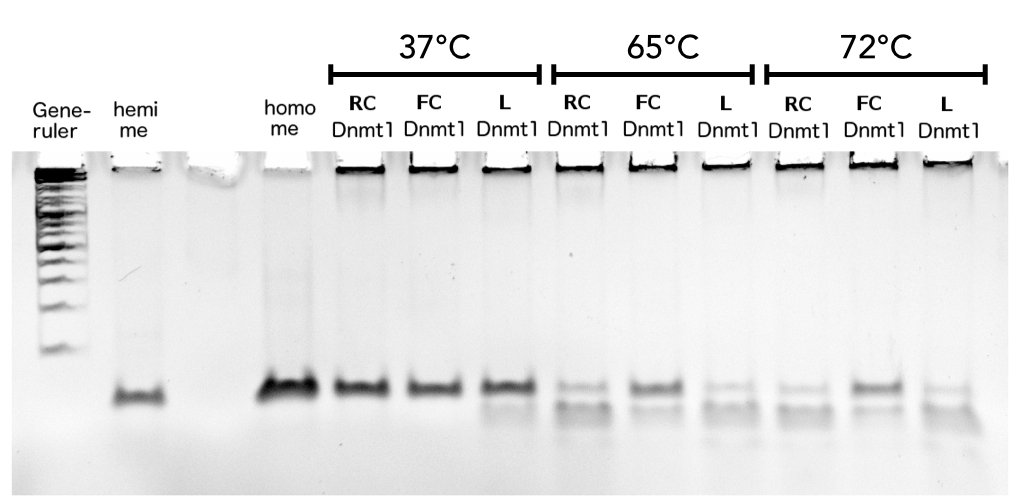
Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 65°C and 72°C.Methylation after one hour was measured. A darker upper band for FC is even visible by eye, indication higher methylation activity.
Discussion
References
[1] Watanabe, M., Yuzawa, H., Handa, N., & Kobayashi, I.. Hyperthermophilic DNA methyltransferase M.PabI from the archaeon Pyrococcus abyssi. Applied and Environmental Microbiology, 72(8), 5367–75 (2006).
[2] Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–70 (2008)
[3] Vieille, C. & Zeikus, G. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65, 1–43 (2001).
[4] Antos, J. M., Popp, M. W.-L., Ernst, R., Chew, G.-L., Spooner, E., & Ploegh, H. L.. A straight path to circular proteins. The Journal of Biological Chemistry, 284(23), 16028–36 (2009).
[5] Tam, J. P. & Wong, C. T. T. Chemical synthesis of circular proteins. J. Biol. Chem. 287, 27020–5 (2012).
[6] Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005).
[7] Jaenisch, R. & Bird, A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33 Suppl, 245–54 (2003).
[8] Ehrlich, M.. Amount and distribution of 5-methycytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 10: 2709–2721 (1982).
[9] Lister, R. & Ecker, J. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 959–966 (2009).
[10] Bird, A.. DNA methylation patterns and epigenetic memory. Genes & Development 16, 6-21 (2002).
[11] Jaenisch, R., and Bird, A.. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33, 245-254 (2003).
[12] Weichenhan, D., and Plass, C.. The evolving epigenome. Human Molecular Genetics 22, R1 (2013).
[13] Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005).
[14] Jurkowska, R. Z., Jurkowski, T. P. & Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 12, 206–22 (2011).
[15] Song, J., Teplova, M., Ishibe-Murakami, S. & Patel, D. J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 335, 709–12 (2012).
[16] Bashtrykov, P. et al. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem. Biol. 19, 572–8 (2012).
[17] Lee, C.-D., Sun, H.-C., Hu, S.-M., Chiu, C.-F., Homhuan, A., Liang, S.-M., Wang, T.-F. An improved SUMO fusion protein system for effective production of native proteins. Protein Science : A Publication of the Protein Society, 17(7), 1241–8 (2008).