"
"
Team:Oxford/biosensor characterisation






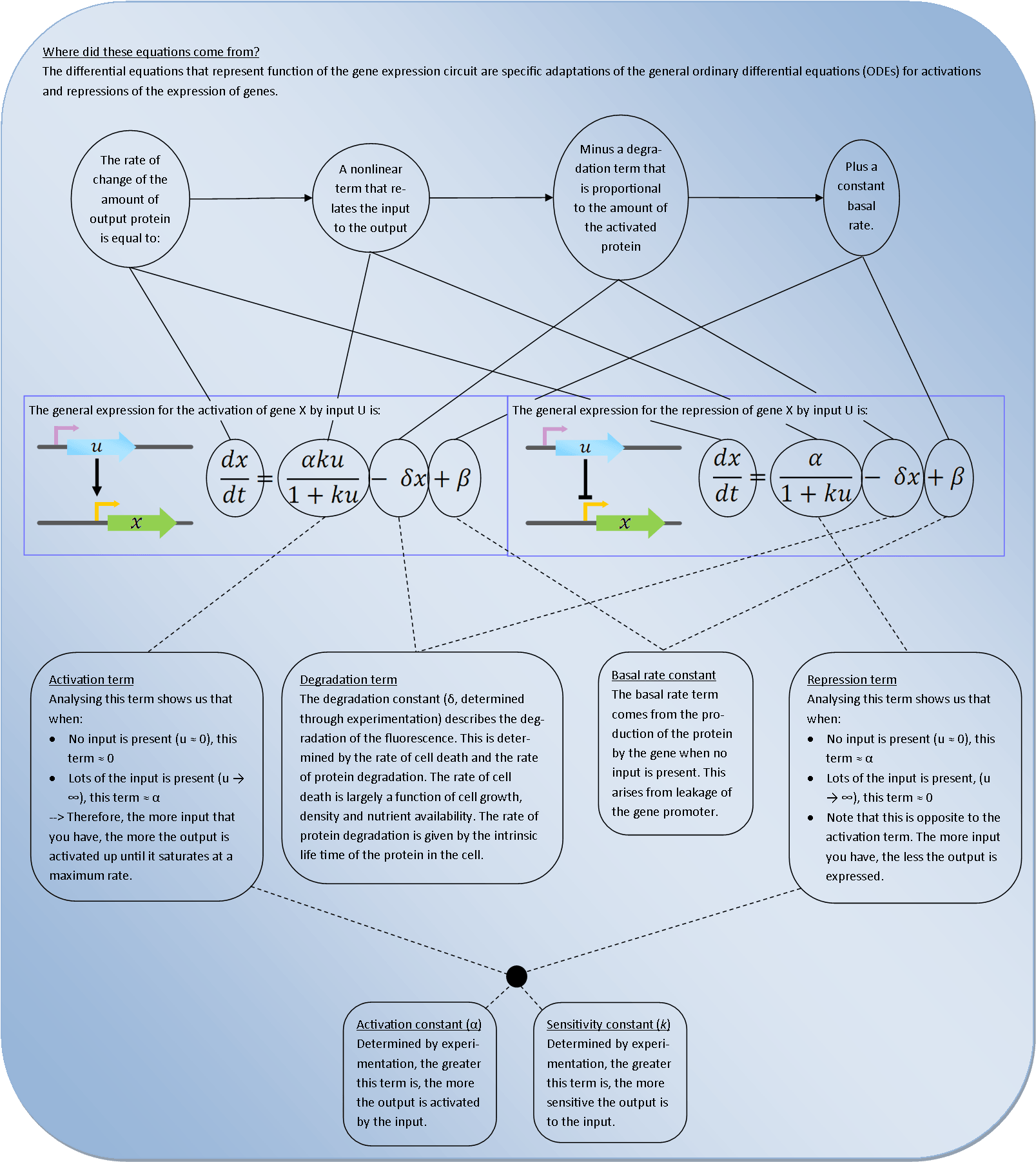
 Deterministic models are very powerful tools for synthetic biology. They describe the behaviour of the bacteria at the population level and use Ordinary Differential Equations (ODEs) to relate each activation and repression. By constructing a cascade of differential equations one can build a realistic model of the average behaviour of the system.
Deterministic models are very powerful tools for synthetic biology. They describe the behaviour of the bacteria at the population level and use Ordinary Differential Equations (ODEs) to relate each activation and repression. By constructing a cascade of differential equations one can build a realistic model of the average behaviour of the system.


 Oxford iGEM 2014
Oxford iGEM 2014





Wetlab data showing response in level of mCherry expressed with different concs of ATC
By making a translational fusion of mCherry at the C terminus of the dcmR gene under the tet promoter and tet operator system (see our Construction page for details) we could measure mCherry fluorescence to gain information about dcmR induction by ATC. Expression was induced with various amounts of ATC and the following fluorescence data acquired. Exposure time was 0.2 seconds. As no calibration data was obtained using purified mCherry, the results have been left in fluorescence arbitrary units. Images were analysed using imageJ software.mCherry fluorescence increases with amount of ATC used confirming that the dcmR gene was expressed under the control of the tet promoter and operator system.






This data was then used to refine and test our models (see below).

Introduction
As you can see from the biochemistry bubble above, our team was only able to obtain fluorescence data for the first half of the genetic circuit (ATC-induced mCherry response). On top of this, the wet lab team were unable to obtain data that measured how the fluorescence of a single culture changed with time, again because of time constraints. This is slightly limiting because it means that we don’t have any dynamic data for any part of our system, and therefore can’t test the modelling predictions of the speed of the biosensor’s response.The original data is shown on the right with error bars showing the standard error of the measurements.
Standard error is calculated as the average standard deviation divided by the square root of the total number of readings.
How we used the model
However, to demonstrate the power of the computer models that we’ve built, we made our model simulate the same graph (mean fluorescence against ATC concentration added). To build this, we started from the graph shown in the first modelling bubble on this page, shown here with a small basal rate (see where did these equations come from? ). This graph shows how the predicted fluorescence of the cells changes with time in response to an addition of ATC halfway along the time scale. At this stage, all input values, model parameters and therefore results are arbitrary.
We then ran the model for the correct amount of time (2 hours 20mins incubation with ATC) and ran it for lots of different concentrations of ATC over the range that the wet-lab team tested. The parameters are still arbitrary at this point (the same as above) and the results of the graphs are therefore arbitrary are as well, but the input values are now correct. The graphic below shows how we used the existing model to obtain the same graphs as the wet-lab team had obtained.
The numerical inputs that were used to model this data set were therefore:

Oxford iGEM 2014
Parameters
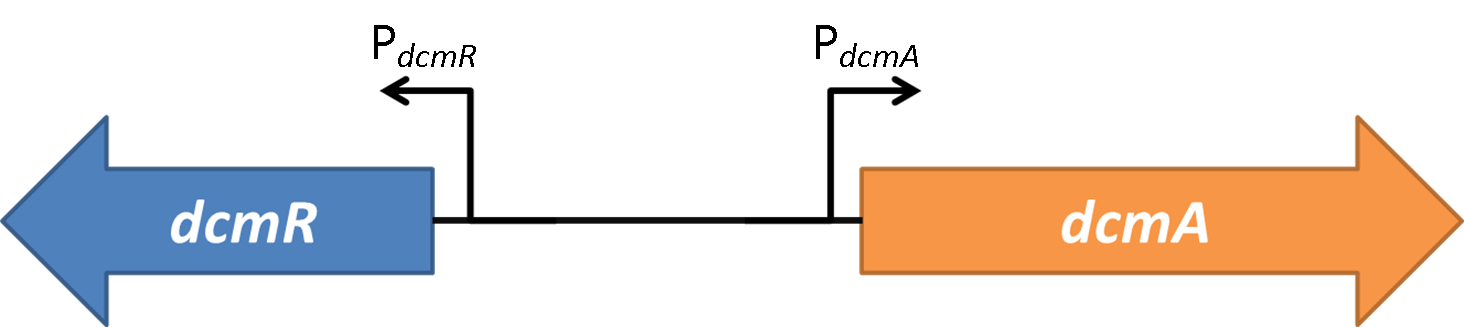
As all results are arbitrary up to this point, it is now time to calculate the parameters that will make the model’s response match up with the wet-lab data. The purpose of doing this is that the model will be able to give relatively accurate predictions of the response of the bacteria to further testing, therefore making the development of the biosensor much more efficient. The amount of data here will not allow us to calculate the parameters to a high level of accuracy, but it should be able to give us some very good approximations of what we can expect. The parameters that we need to calculate are the constants in the differential equation that governs the behaviour of the first half of the genetic circuit. This half of the system is shown again here to remind the reader which part we are considering.
The parameters that we need to calculate are the constants in the differential equation that governs the behaviour of the first half of the genetic circuit. This half of the system is shown again here to remind the reader which part we are considering.
These parameters are:
Remember that because the mCherry gene is tagged (translational fusion) onto the end of the dcmR gene, the mCherry fluorescence will be the same as the amount of DcmR protein present. However, there is not very comprehensive data in the literature about the values that we can expect from the behaviour of the dcmR gene and its stability in vivo.
Degradation constant
The initial steady state of the system (before ATC has been added) is determined by two constants in the model. These constants are the degradation constant of DcmR and the basal transcription rate of the system. Due to the lack of numerical information in the literature on the behaviour of the dcmR gene, the way of calculating these two parameters is by using the single basal rate data point from the wet-lab data (fluorescence value when 0ng of ATC has been added).If we assume that the half-life of the dcmR protein is 3 hours [1] (180 minutes), we can calculate the degradation constant for our model. The exponential protein decay is therefore described by:

Basal transcription rate
At the basal steady state, the rate of change of DcmR (and therefore fluorescence) is zero. As no ATC has been added to the system yet, the value of [ATC] is also zero. This simplifies the equation to:
This simplifies the equation to:
 As we want our model to accurately predict the fluorescence, we will substitute the fluorescence value in place of the [DcmR] and rearrange:
As we want our model to accurately predict the fluorescence, we will substitute the fluorescence value in place of the [DcmR] and rearrange:
 Substituting in the value for δ1 that we found above and the basal steady state fluorescence level from the data (471 to 3 s.f.) gives the basal transcription rate as:
Substituting in the value for δ1 that we found above and the basal steady state fluorescence level from the data (471 to 3 s.f.) gives the basal transcription rate as:

Expression rate constant and Michaelis - Menten constant
After the ATC has been added to the system, the value of [ATC] becomes non-zero. This means that the expression constant and the Michaelis – Menten constant start to affect the system.To find the parameters that make the model’s output match the data values, we turned to the code that we had developed for parameter calibration.
How are we calculating the parameters?
This code gave the parameters as:
α1 = expression rate constant of dcmR = 16.5 (fluorescence/min)
k1 = Michaelis - Menten constant of dcmR = 0.015 (ml/ng)
Entering the correct parameters
When the parameters that had been calculated above were entered into the model: alongside the correct inputs:
alongside the correct inputs:
 The graph below shows the model's predictions plotted in the same figure as the data points that the wet-lab team obtained for the system:
The graph below shows the model's predictions plotted in the same figure as the data points that the wet-lab team obtained for the system:
 Plotting the model's output as a by interpolating between the calculated values makes the graph clearer:
Plotting the model's output as a by interpolating between the calculated values makes the graph clearer:

Sensitivity
An important part of building mathematical models is sensitivity analysis of the results. This can be basically explained as wiggling all of the input values and parameters to see how much variations in each of these values affects the system output. This is especially important for finding parameters to describe the system as it is important to know what level of accuracy the values need to be found to provide a reasonable degree of prediction accuracy.On top of this, it is possible to find what range of values the system is especially sensitive to. An example of this analysis is shown with a simple example that is relevant to our system below:

Stability
In our Engineering studies we have learnt detailed control theory. Control theory is an interdisciplinary branch of engineering and mathematics that deals with the behaviour of dynamical systems with inputs, and how their behavior is modified by feedback. The usual objective of control theory is to control a system so that its output follows a desired control signal, called the reference, which may be a fixed or changing value. This important because many dynamic systems can go unstable if they are given an unsafe set of input values and/or operating conditions.However, as there are no feedback loops in this synthetic circuit, control theory analysis of this system isn't necessary.
Future experiment ideas from an Engineering design perspective
To see details about how the wet lab team then used this model to guide their work see our optimisation page.Reference
[1] Dr George Wadhams by personal communication (14/10/2014)
The first data readings that we managed to obtain from the completed genetic circuit were qualitative sfGFP fluorescence readings. The results of these are shown here:
 The first system that we tested is shown here. This is the bottom level of our synthetic circuit. Testing just this plasmid allowed us to obtain some important information to allow us to characterise the genetic system.
The first system that we tested is shown here. This is the bottom level of our synthetic circuit. Testing just this plasmid allowed us to obtain some important information to allow us to characterise the genetic system.
The GFP fluorescence of this system was high and this is the basal transcription rate of the PdcmA promoter.
 The second system that we tested was the whole synthetic system without any DCM added. This allowed us to analyse just the effect of DcmR on the PdcmA promoter when it was compared to the result from system 1.
The second system that we tested was the whole synthetic system without any DCM added. This allowed us to analyse just the effect of DcmR on the PdcmA promoter when it was compared to the result from system 1.
The fluorescence was much lower than the basal transcription rate of the PdcmA. This strongly suggests that the DcmR protein acts as a repressor of the PdcmA.
The next step is to prove that adding DCM represses this repression and increases the sfGFP fluorescence of the system.
 Therefore we know that DcmR represses PdcmA with DCM repressing this repression. This means that the system is a double repressor!
Therefore we know that DcmR represses PdcmA with DCM repressing this repression. This means that the system is a double repressor!
First system tested
The first system that we tested is shown here. This is the bottom level of our synthetic circuit. Testing just this plasmid allowed us to obtain some important information to allow us to characterise the genetic system.
The GFP fluorescence of this system was high and this is the basal transcription rate of the PdcmA promoter.
Second system tested
The second system that we tested was the whole synthetic system without any DCM added. This allowed us to analyse just the effect of DcmR on the PdcmA promoter when it was compared to the result from system 1.
The fluorescence was much lower than the basal transcription rate of the PdcmA. This strongly suggests that the DcmR protein acts as a repressor of the PdcmA.
The next step is to prove that adding DCM represses this repression and increases the sfGFP fluorescence of the system.
Third system tested
Therefore we know that DcmR represses PdcmA with DCM repressing this repression. This means that the system is a double repressor!






Oxford iGEM 2014