 "
"
Team:Heidelberg/pages/Toolbox
From 2014.igem.org
Jelisaweta (Talk | contribs) (→Results) |
Jelisaweta (Talk | contribs) (→Results) |
||
| Line 236: | Line 236: | ||
part7 <br /> | part7 <br /> | ||
part8 <br /> | part8 <br /> | ||
| - | + | ||
| - | + | ||
<html> | <html> | ||
Revision as of 00:02, 18 October 2014
A primary mission of the iGEM team Heidelberg 2014 is to provide a new toolbox based on appropriate standards that introduces the highly functional INTEINS to the parts registry and thereby paving the way to a new level of posttranslational modification possibilities for the iGEM community. Inteins are self-excising peptide segments that mediate the reaction of protein trans-splicing. They don't require any additional energy sources to perform the splicing reaction. Another valuable feature of inteins is their specific recognition, also in the presence of other proteins, which makes them functional in vitro as well as in vivo. On this page we describe experimental approaches to provide and evaluate five related but still quite different applications using inteins and their splicing capabilities. Thereby we present scientific prove of the diversity and efficiency of an intein toolbox that can be used for a wide range of posttranslational modifications of proteins. Explore the new possibilities, intein usage provides! On this page we describe the scientific work in form of modeling and experiments that lead to the establishment of the first intein toolbox in the history of iGEM!
Jump right into splicing and use the toolbox guide to design your intein-equipped proteins of choice!





Contents |
Circularization
Millions of years of evolution have allowed proteins to performed extremely specific chemical modifications that are not only essential for living organisms but can also be of great benefit to produce useful molecules for our life, efficiently and at low cost. A major limitation of the use of enzyme for industrial application and in general for usage out of their natural environment is their stability. They can be destroyed by other enzymes and they can unfold and take non-functional conformation when exposed to non-physiological temperature and pH. Such limitations has motivated research in species that can grow at extreme temperatures [6]. Another major area of chemical research if the design of strategies to stabilize enzymes, and more generally proteins and peptides. Protein circularization, meaning ligation of the N- and C-terminal ends of a protein, represents a promising way to achieve this stabilization. While conserving the functionality of their linear counterpart, circular proteins can be superior to linear proteins in terms of thermostability [1][2][3], resistance against chemical denaturation [4] and protection from exopeptidases [2][4]. Moreover, a circular backbone can improve in vivo stability of therapeutical proteins and peptides [5]. These remarkable properties motivated us to develop new tools to circularize any protein of interest.
Our Toolbox Guide provides a step-by-step strategy to clone a circularization linker and express it in e. Coli. Moreover, in case of complex structures where the protein extremities are far from each other, we have developed a software that will design the appropriate rigid linkers.
Please find more information and results of the experiments that laid the foundation for the circularization kit of the toolbox on the Circularization pages.
Oligomerization
Split inteins constitute a useful tool to produce huge polymers in vivo: Hauptmann et al. managed to fabricate synthetic spider silk with microfiber structure. The results using an easy-to-handle split intein system were stunning: The polymers had a molecular weight of 250 kDa and more [7].
Standardization of oligomerization
The valuable properties of spider silk, for example its exceptional strength and elasticity, result from numerous repeats of certain protein motifs. Convenitonal methods to multimerize these motifs bear a lot of difficulties: Often genetic and mRNA instability constitute a barrier for the production of multimers as fusion proteins.[1] Posttranslational assembly through split inteins is therefore the solution to overcome these problems. The successfull polimerization of spider silk potein motifs demonstrates the potential of split inteins to be a useful tool for the production of new biomaterials by performing oligomerization reactions with split inteins. The iGEM team Heidelberg standardized (lik to toolbox guide) the oligomerization procedure with split inteins to allow easy handling with different proteins.
The mechanism

Circularization is achieved by bringing the N and C terminus of a protein very close together, so both intein parts can asseble, cut out off the protein and thereby circularize it. In contrast, oligomerization occurs when the both termini of a protein cannot reach each other and the intein parts of two neighbouring proteins assemble, at the same assembling the protein parts.
Fusion and Tagging
Post-translational modifications are highly prevalent in nature: Almost every protein in a cell is modified after being translated, numerous varieties of the protein are formed from the mere protein backbone. Synthetic Biology, however, has not yet made use of the innumerable possibilities nature has developed. With our collection of intein assembly constructs we expand the arsenal of synthetic biology by enabling unlimited changes of a protein's amino acid sequence even after translation.
To be able to fuse any two halves of a protein together can have many different uses. We therefore saw the need for a standardised construct, the intein assembly part. This BioBrick part allows the user to clone two DNA-sequences coding for two parts of a peptide into a plasmid prepared with selection markers and standardised overhangs. Those parts were all send in with additional hexahistidine-tags to enable quick analysis on a western blot, however there are highly customisable parts available as well. In an extensive assay we proved the principle behind split protein assembly by showing that GFP can be artificially split into two halves and thereafter be reassembled so the fluorescence is restored. Visit the split Fluorescent Protein
Applying the Tagging Tool allows for all kinds of natural as well as synthetic posttranslational modifications by the chemoselective addition of a peptide to a recombinant protein. The principle is based on intein-mediated protein fusion using the SspDnaB split intein for N-terminal modifications and SspDnaX-S11 for C-terminal ones. The split SspDnaB intein has a very short N-terminal part, consisting of only 11 amino acids intein, 5 amino acids extein sequence and a much longer C-terminal part. By contrast, SspDnaX-S11 has a C-terminal part consisting of only 6 amino acids intein and 3 amino acids extein sequence, but a much longer N-terminal part. The short part including the desired modification is easy to obtain by chemical synthesis. This offers the possibility to introduce this modification at a specific locus. There are different publications on intein-based introduction of posttranslational modifications, including phosphorylation, lipidation, glycosylation, acetylation and ubiquitination [A]. Phosphorylation, for example has been applied with tyrosine kinase C-terminal Src kinase (Csk) in order to be able to study the structure and function of this specifically modified protein [B]. In general, split inteins are powerful tools to easily introduce post-translational modifications in a chemoselective manner and with the intein fusion and tagging parts this tool becomes easily available to many people.
Posttranslational Modifications
Posttransplational modifications are highly prevalent in nature: Almost every protein in a cell is modified after having been translated, adding numerous varieties of the protein to the mere protein backbone. Synthetic Biology, however, can expand the possibilities offered by nature and introduce synthetic posttranlational modifications or attachments. Applying the intein assembly constructs the iGEM team Heidelberg provides a tool for all kinds of natural as well as synthetic posttranslational modifications by chemoselective addition of a peptide to a recombinant protein. The principle is based on intein-mediated protein fusion using the SspDnaB split intein for N-terminal modifications and SspDnaX-S11 for C-terminal ones. The split SspDnaB intein has a very short N-terminal part, consisting of only 11 amino acids intein, 5 amino acids extein sequence and a much longer C-terminal part. By contrast, SspDnaX-S11 has a C-terminal part consisting of only 6 amino acids intein and 3 amino acids extein sequence, but a much longer N-terminal part. The short part including the desired modification is easy to obtain by chemical synthesis. This offers the possibility to introduce this modification at a specific locus. There are different publications on intein-based introduction of posttranslational modifications, including phosphorylation, lipidation, glycosylation, acetylation and ubiquitination [A]. Phosphorylation, for example has been applied with tyrosine kinase C-terminal Src kinase (Csk) in order to be able to study the structure and function of this specifically modified protein [B]. In general, split inteins are powerful tools to easily introduce posttranslational modifications in a chemoselective manner, Use our toolbox guide to attach posttranslational modifications and attachments to your protein of interest!
Translocation
Regulated targeting of proteins to certain loci inside the cell can be achieved using the iGEM Heidelberg 2014 translocation tags for either E. coli or eucaryotes. We provide following tags:
Use our toolbox guide to add or remove iGEM Heidelberg translocation or custom tags to your protein of interest!
Regulated targeting of proteins to certain loci inside the cell can be achieved using the iGEM Heidelberg 2014 translocation tags for either E. coli or eukaryotic expression systems.
| E. coli: MinD membrane targeting sequence (BBa_K1362055) | → membrane localization |
| OmpA (BBa_K1362060) | → surface display |
| pelB leader sequence (BBa_K1362058) | → periplasmic localization |
| Eucaryotic: myristooilation signal sequence (BBa_K1362056) | → membrane localization |
| PKI-NES (BBa_K1362059) | → cytoplasmic localization |
| NLS (BBa_K1362057) | → nuclear localization |
Use our toolbox guide to post-translationally assemble proteins, attach tags or change the location of your protein!
Cloning strategy
All translocation tags except from the OmpA surface protein tag were ordered as DNA oligos containing the tag sequence, extein sequence and BsaI oberhangs. OmpA surface protein was obtained from the Spring Distribution 2012 and also PCR amplified with overhangs. By Golden Gate Assembly the tags were cloned into the carrier constructs BBa_K1362052 (for C-terminal tags) and BBa_K1362053 (for N-terminal tags). These carrier constructs, pSB1C3 backbones with two BsaI sites as an insert, were used to send the tags to the registry of standard biological parts. The user can afterwards easily use Golden Gate Assembly to clone the tags from the carrier constructs into intein assembly constructs.
Results
The parts were sequenced, the results were positive. You can obtian them here:
MinD MTS:BBa_K1362055
pelB leader sequence: BBa_K1362058
myristoilation signal sequence: BBa_K1362056
PKI-NES: BBa_K1362059
NLS: BBa_K1362057
OmpA (part1, part2 , part3 , part4 ): BBa_K1362060
Purification
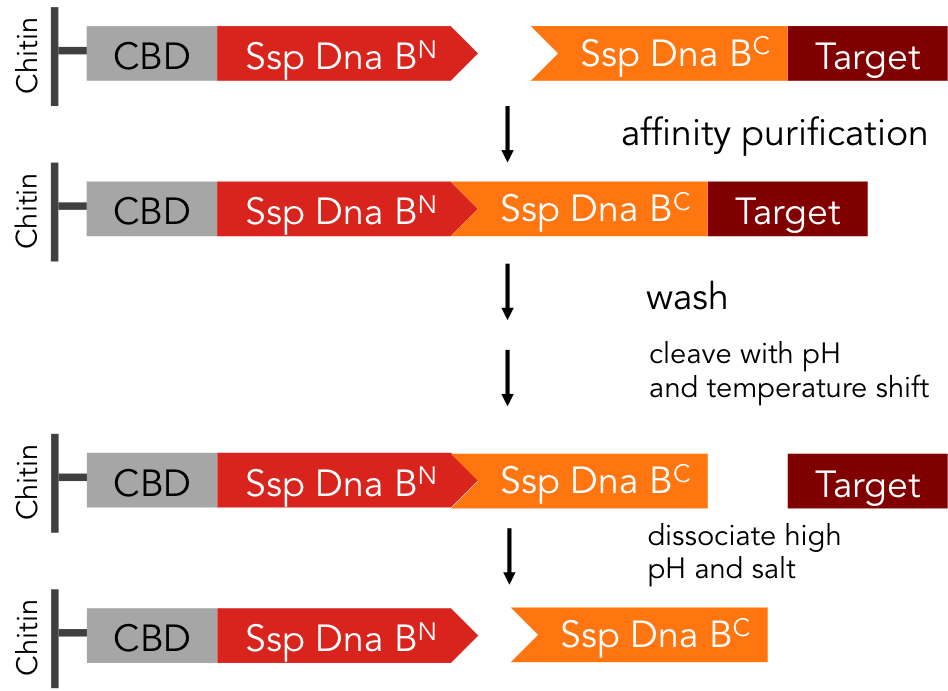
In previous purification procedures, there a several steps to take to get solely the purified protein: The first step is the binding of the protein to the affinity matrix, the washing and finally release of the protein from the affinity column, the protein still being bound to the affinity tag. The next step is the removal of the affinity tag from the protein by protease cleavage, which causes undesirable secondary cleavage within the protein sequence. In industrial scales proteases are the most expensive component during the purification process. Finally, the split off affinity tag has to be separated from the protein, which takes again a lot of effort.
The split-intein based system is manifold easier: All these steps can be combined into one by using Ssp DnaB mini-intein, an artificial split intein. It performs the trans-splicing reaction only under certain pH and temperature conditions. A mutation of the SspDnaB N-intein at Cys1 to Ala prevents cleavage at the N-terminus, cleavage at the C-terminus still occurs. The N-terminal intein part, attached to a chitin binding domain can be used as an affinity ligand, while the C-terminal part, fused to a protein of interest, can be used as an affinity tag binding highly specific to the ligand part. After loading the protein of interest on a chitin column, the trans-splicing reaction can be induced by adjusting temperature and pH: the both split inteins fuse together, disrupting the fusion to the protein of interest in one single step [C].
On Off
On
Off

§§§§§§
§§§§§§
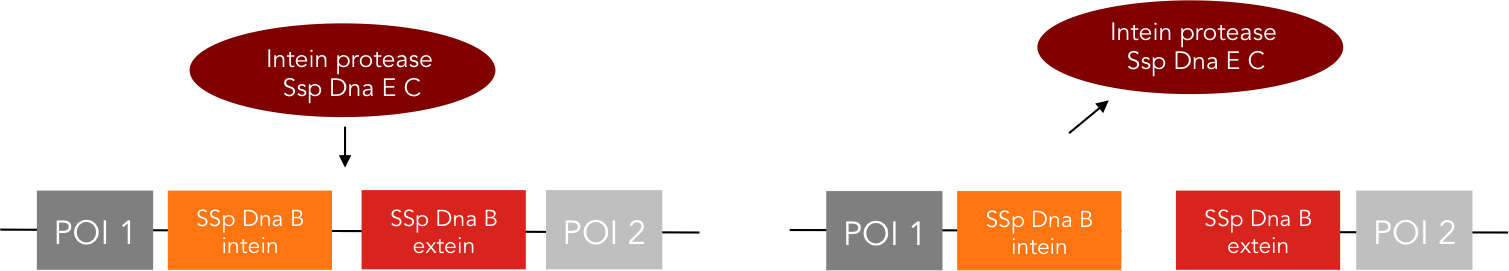
We provide two different tools for deactivation of proteins. One of them is the C-terminal degradation tag SsrA, the other one the intein protease.The SsrA tag directs the tagged protein to the ClpXP protease, causing degradation of this protein (1). The intein protease is a singular tool for site-specific in vivo protein cleavage. Under in vitro conditions, protease usage is relatively easy compared to in vivo conditions. The inside of a cell constitutes a very complex environment, so that conventional proteases would simply cleave not only the desired site of the protein of interest, but also other proteins inside this cell, causing huge interference with pathways crucial for cell functions. So far, only the tobacco etch virus (TEV) protease has been used with success for in-vivo cleavage without severe decline of cell viability.(3) Volkmann et al. first showed in vitro functionality of the intein protease. (2) Later it was shown, that the intein proteease is also applicable in vivo in E. coli and yeast cells without impairment of cell functions. (3)
The intein protease consists of the 144-aa SspDnaB S1 C-intein (with an Asn-to-Ala mutation at the end of the C-intein to prevent protein splicing) followed by the extein sequence. This C-terminal part of the split intein recognizes its counterpart, the N-terminal split intein inserted into the sequence of the proteine to be cleaved, and cleaces there between extein and intein sequence.
Intein protease cloning strategy
SspDnaB C intein was PCR amplified from a plasmid (pCL20) received from Prof. Dr. Henning Mootz. The PCR primers contained overhangs to get the following constuct:
XbaI site – T7RBS - His6 – SspDnaB C intein (N → A) – SspDnaB C extein – SpeI site – PstI site
This PCR product and pSB1C3 were digested with XbaI and PstI. The digested PCR product was ligated into pSB1C3. The resulting plasmid is BBa_K1362050 (Intein protease – T7RBS-His-SspDnaBC ).
The next step was to add a promotor. Therefore, BBa_K1362050 was digested with EcoRI and XbaI. BBa_K808000, an Ara promotor, was digested with SpeI and PstI. These two parts were ligated, resulting in BBa_K1362051 (Intein protease with arabinose inducible regulatory promoter/ repressor araC-Pbad-T7RBS-His-SspDnaBC).
Results
Both intein protease constructs BBa_K1362050 and BBa_K1362051 were sequenced. We obtained positive results. Here you can see the sequencing results of BBa_K1362051, the longer of the constructs (and including BBa_K1362050),
BBa_K1362051
part 1
part2
part3
part4
part5
part6
part7
part8
SsrA degradation tag cloning strategy
The SsrA degradation tag was ordered as DNA oligos containing the tag sequence, extein sequence and BsaI overhangs. By Golden Gate Assembly the tag was cloned into the carrier construct BBa_K1362052 (for C-terminal tags). This carrier construct consists of a pSB1C3 backbone with two BsaI sites as an insert. It was used to send the tag to the registry of standard biological parts. The user can afterwards easily use Golden Gate Assembly to clone the tag from the carrier constructs into intein assembly constructs.
Results
The part was sequenced, the results were positive. You can see the sequencing results here:
SsrA: BBa_K1362054
References
[1] McGinness, KE et al.: Engineering controllable protein degradation. Molecular Cell 22, 701–707, June 9, 2006. DOI 10.1016/j.molcel.2006.04.027
[2] Volkmann, G. et al: Controllable protein cleavages through intein fragment complementation. Protein Sci, 18 (2009), pp. 2393–2402. DOI: 10.1002/pro.249
[3] Volkmann, Gerrit et al.: Site-specific protein cleavage in vivo by an intein-derived protease. FEBS Letters 586 (2012) 79–84. doi:10.1016/j.febslet.2011.11.028.
[7] Hauptmann, V. et al.: Native-sized spider silk proteins synthesized in planta via intein-based multimerization. Transgenic Res (2013) 22:369–377. DOI 10.1007/s11248-012-9655-6.
[A] Vila-Perello, Miquel et al.: Biological Applications of Protein Splicing. Cell 143. October 15, 2010. DOI 10.1016/j.cell.2010.09.031.
[B] Muir, Tom W. et al.: Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. USA 95 (1998).
[C] Lu, Wei et al.: Split intein facilitated tag affinity purification for recombinant proteins with controllable tag removal by inducible auto-cleavage. J. Chromatogr. A 1218 (2011)
[D] Vila-Perello, Miquel et al.: Biological Applications of Protein Splicing. Cell 143. October 15, 2010. DOI 10.1016/j.cell.2010.09.031.
[E] Muir, Tom W. et al.: Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. USA 95 (1998).
(1) McGinness, KE et al.: Engineering controllable protein degradation. Molecular Cell 22, 701–707, June 9, 2006. DOI 10.1016/j.molcel.2006.04.027
(2) Volkmann, G. et al: Controllable protein cleavages through intein fragment complementation. Protein Sci, 18 (2009), pp. 2393–2402. DOI: 10.1002/pro.249
(3) Volkmann, Gerrit et al.: Site-specific protein cleavage in vivo by an intein-derived protease. FEBS Letters 586 (2012) 79–84. doi:10.1016/j.febslet.2011.11.028.