 "
"
Team:SUSTC-Shenzhen/Project
From 2014.igem.org
| Line 66: | Line 66: | ||
===Plasmid & experiment design=== | ===Plasmid & experiment design=== | ||
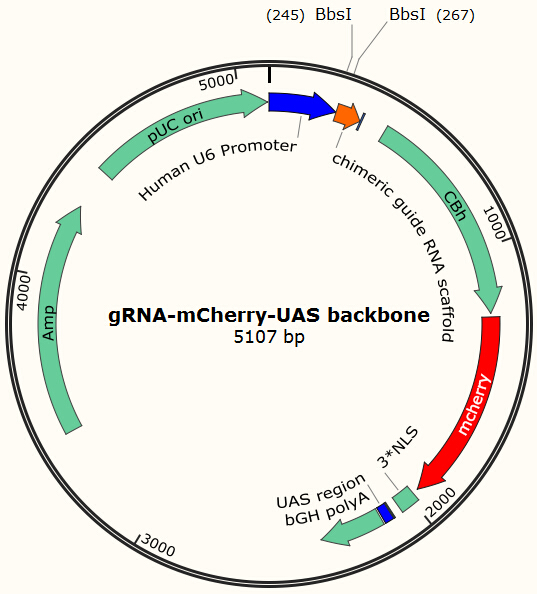
Before using this delivery system, we need to check the delivery efficiency of this protein system at first. So, firstly, we construct a plasmid that carries a sgRNA for EGFP, two GAL4 sequences which are necessary for the vehicle’s recognition and binding and a mCherry gene sequence followed by a NLS (nucleus leading sequence). Figure 9 demonstrates the plasmid we construct. | Before using this delivery system, we need to check the delivery efficiency of this protein system at first. So, firstly, we construct a plasmid that carries a sgRNA for EGFP, two GAL4 sequences which are necessary for the vehicle’s recognition and binding and a mCherry gene sequence followed by a NLS (nucleus leading sequence). Figure 9 demonstrates the plasmid we construct. | ||
| - | + | <center>{{SUSTC-Image|wiki/images/b/b2/SUSTC-Shenzhen-Project-gRNA-mCherry-UAS-backbone.jpg}}</center> | |
| - | Figure 9 Constructed plasmid to deliver gRNA into host cell based on pX330 backbone | + | <center>Figure 9 Constructed plasmid to deliver gRNA into host cell based on pX330 backbone</center> |
| + | |||
The construction is based on the plasmid backbone of Zhang Lab’s pX330. There are two BbsI restriction enzyme sites between U6 strong promoter and U6 terminator in which we can insert sgRNA. Once sgRNA is inserted, the two BbsI sites disappear and the sgRNA can’t be changed anymore. Figure 10 shows how sgRNA replacement is blocked once BbsI sites are used. | The construction is based on the plasmid backbone of Zhang Lab’s pX330. There are two BbsI restriction enzyme sites between U6 strong promoter and U6 terminator in which we can insert sgRNA. Once sgRNA is inserted, the two BbsI sites disappear and the sgRNA can’t be changed anymore. Figure 10 shows how sgRNA replacement is blocked once BbsI sites are used. | ||
| - | + | ||
Figure 10 Mechanism of gRNA insertion into pX330-derived plasmid backbone | Figure 10 Mechanism of gRNA insertion into pX330-derived plasmid backbone | ||
CBh is a eukaryotic promoter, followed by mCherry and 3*NLS. NLS makes red fluorescent protein enter into cell nucleus after expression. By means of PCR and molecular cloning, we can change the number of UAS, a sequence of 17bp which is recognized and bound by GAL4 protein. The sgRNA we used in this plasmid is for EGFP, to collaborate with the Cas9 system and EGFP gene that we have integrated into Hela cell line. In this way, we can visualize how many plasmids are transformed into nucleus by detecting the quantity of red-fluorescence-emitting cells. And with the ratio of reduced green fluorescence and the quantity of red fluorescence we can roughly obtain the efficiency of the CRISPR-Cas system we created. And finally, we reconstruct the plasmid but this time with the sgRNA targeting HIV and test its efficacy. | CBh is a eukaryotic promoter, followed by mCherry and 3*NLS. NLS makes red fluorescent protein enter into cell nucleus after expression. By means of PCR and molecular cloning, we can change the number of UAS, a sequence of 17bp which is recognized and bound by GAL4 protein. The sgRNA we used in this plasmid is for EGFP, to collaborate with the Cas9 system and EGFP gene that we have integrated into Hela cell line. In this way, we can visualize how many plasmids are transformed into nucleus by detecting the quantity of red-fluorescence-emitting cells. And with the ratio of reduced green fluorescence and the quantity of red fluorescence we can roughly obtain the efficiency of the CRISPR-Cas system we created. And finally, we reconstruct the plasmid but this time with the sgRNA targeting HIV and test its efficacy. | ||
Revision as of 04:05, 15 October 2014
Project Description
What is our story?
Abstract
In this project, we want to establish a more effective HIV-curing system with less side-effects by integrating CRISPR/Cas system into human hematopoietic stem cells, which aims to protect the helper T cells from virus infection. The gRNA is designed to target the relative conserved regions in HIV viral genome and inactivate its biological activity. Since viral vectors seem to be of limited use in gene therapy strategies (e.g., potential pathogenicity), there is still a need for a simple, efficient nucleic acid transfer system which allows the target-cell-specific introduction of nucleic acids. By using non-viral DNA delivery system like A-B-toxin-GAL4 fusion protein, we can deliver plasmids encoding gRNA into the helper T cells and readily attack multiple HIV genome sites simultaneously and update the targets along with our knowledge of HIV.
Background
HIV (Human immunodeficiency virus)
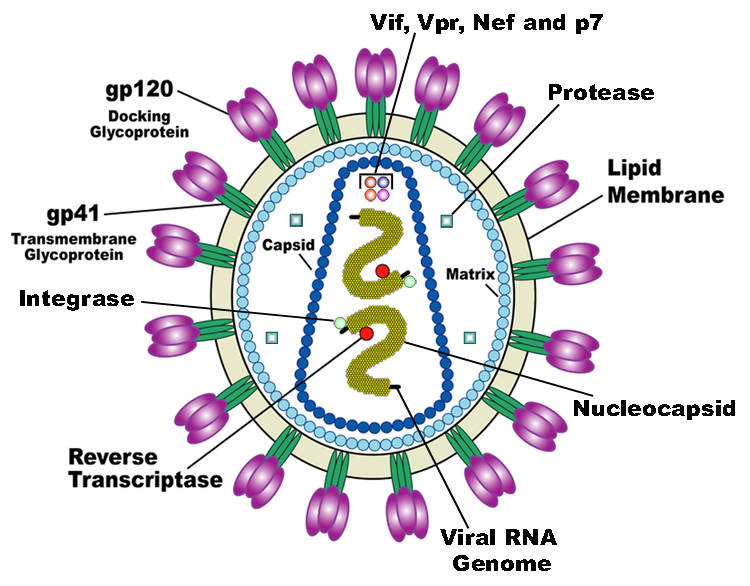
HIV is the trigger of the lethal disease AIDS. It replicates in and kills helper T cells, weakening human immune system and allowing life-threatening opportunistic infection and cancers to thrive. HIV infects vital cells in the human immune system such as helper T cells (especially CD4+ T cells), macrophages, and dendritic cell. As a retrovirus, HIV reverse transcribes its genome and integrated it into the host cell genome after infection. Figure 1 demonstrates the schematic structure of HIV virus (Source: Wikipedia).
Traditional therapies like chemotherapy or radiotherapy can’t protect the helper T cells from virus invasion and the virus can lurk in the helper T cells; drug treatments like HAART (Highly Active Antiretroviral Therapy) can only control the development of the disease and they also have strong side-effects. The process of reverse transcription is extremely error-prone, and the resulting mutations may cause drug resistance or allow the virus to evade the body's immune system.
CRISPR/Cas system
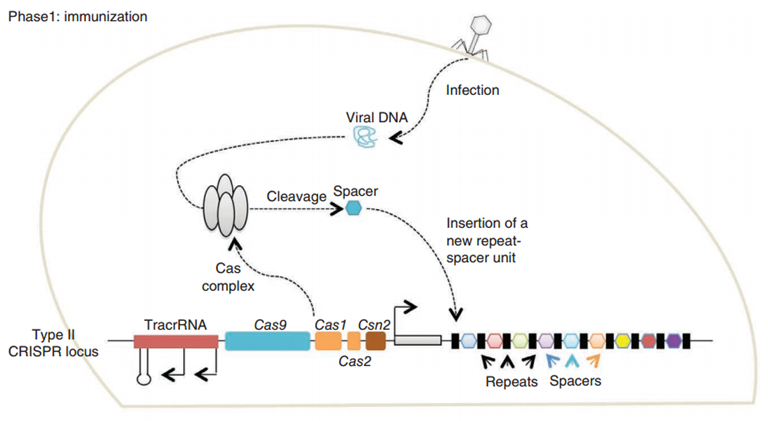
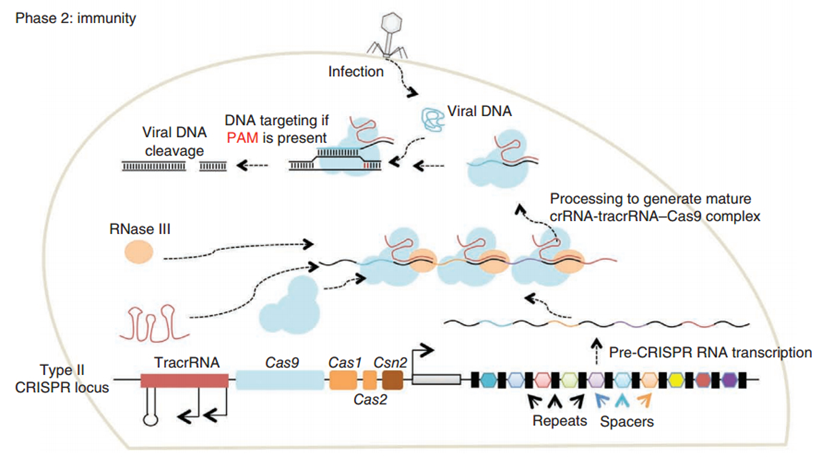
CRISPR/Cas (Clustered regularly interspaced short palindromic repeats/CRISPR-associated system) is originally a self-protecting mechanism in bacteria against external DNA such as virus genome or plasmids. It uses CAS complex to cut the external viral or plasmid DNA and integrating a short DNA segment into the CRISPR loci in the bacteria genome. This short DNA segment will be transcribed into pre-crRNA and bind to another CAS endonuclease called Cas9 with the help of tracrRNA. The Cas9 endonuclease cuts the external DNA in the existence of not only crRNA but also a special recognizing sequence at the 3’ end of the target DNA called PAM (Protospacer Adjacent Motif). When the same external DNA invades next time, the Cas9-tracrRNA-crRNA complex will recognize and cut it in order to destroy its biological activity. Figure 2 demonstrates the mechanism of Type II CRISPR/Cas system in bacteria (Mali, Esvelt, & Church, 2013). Dr. George Church (Mali, Yang, et al., 2013) and Dr. Feng Zhang (Cong et al., 2013) both have successfully transfected Type II CRISPR-Cas system into human cells, which is the foundation of this project.
A-B toxin
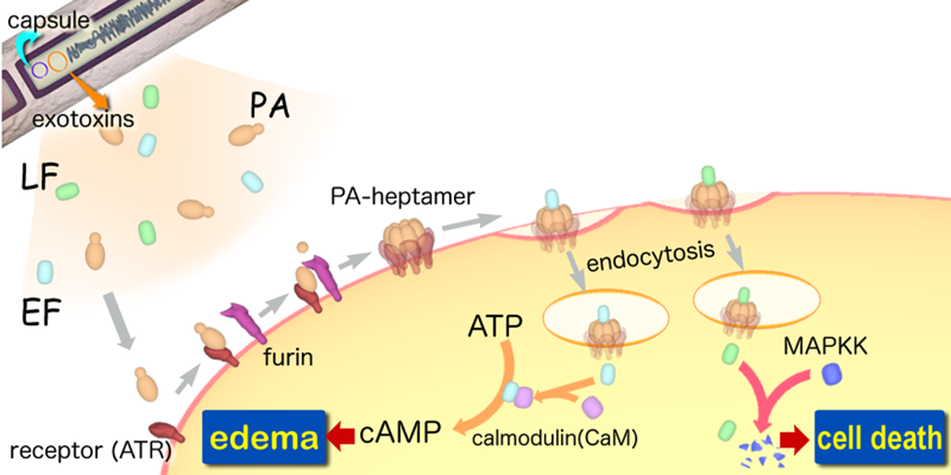
A-B toxin are two-component exotoxin secreted by a number of pathogenic bacteria. The complexes contain two subunits called A subunit and B subunit. A subunit is the active portion that is poisonous to host cells, and is transferred to the host cell through endosomes mediated by the B subunit. The B subunit is a translocation and binding protein. Figure 3 shows how Anthrax toxin enter into human cells.
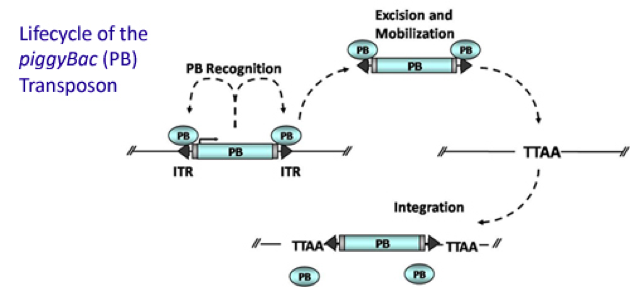
PiggyBac (PB) transposon system
PiggyBac (PB) transposon is a mobile genetic element that efficiently transposes between vectors and chromosomes via a "cut and paste" mechanism but leave no “footprint”. PBase in this system recognizes the transposon-specific inverted terminal repeat sequence (PB5 & PB3 in Figure 6) located on both end of transposon vector and efficiently moves the contents from the original sites and efficiently integrates them into TTAA chromosomal sites. Figure 4 demonstrates the working mechanism of PB transposon system (Source: Wikipedia Author: Transposagenbio).
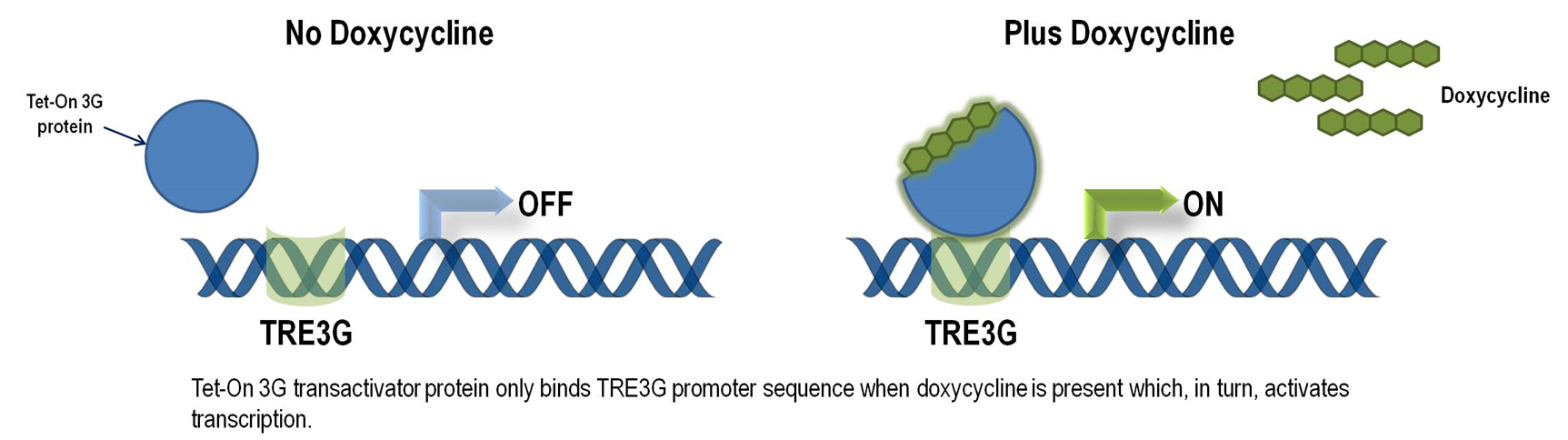
Tet-ON 3G operon system
The Tet-On 3G Systems are inducible gene expression systems for mammalian cells. Target cells that express the Tet-On 3G transactivator protein and contain a gene of interest (GOI) under the control of a TRE3G promoter (PTRE3G) will express high levels of your GOI, but only when cultured in the presence of doxycycline (Dox), which is a synthetic tetracycline derivative. The TRE3G promoter is very sensitive to the concentration of doxycycline and functions like a step function after induction. Also, the Dox concentrations required for induction of Tet-On Systems are far below cytotoxic levels for either cell culture or transgenic studies. So it is ideal to use Tet-ON 3G as a mammalian gene expression controller. Figure 5 demonstrates the mechanism of Tet-ON 3G system (Source: Clontech)
Experiment design
Written by Lin Le, Sijia Liu, Mengqi Xu, Yongkang Long & Yicong Tao
Constructing stable CRISPR/Cas system in human cells
The program aims to endow the human cell the capacity of resisting the infection of HIV by introducing the pruned CRISPR/Cas9 system into the human cell. To wrestle with the high mutation rate of HIV and improve the flexibility of the system, we use PiggyBac transposon to transfect hematopoietic stem cells for permanently and stably expressing Cas9 protein such that all helper T cells differentiate from modified hematopoietic stem cell have ability to defeat HIV by gRNA which is transfected into the cells in need. The DNA segment expressing Cas 9 from pX-330 is cloned into the PiggyBac transposon vector (PB-TetOn-Cas9, Figure 6) which contains a puromycin resistance gene for selection. Cas9 could potentially cause host genome mutagenesis and chromosomal disorders, cytotoxicity, genotoxicity, or oncogenesis. So we use Tet-on 3G operon which is induced by doxycycline to efficiently control the expression of Cas9 in eukaryotic cells and fortify the safety of the system.

To test the efficiency of the Cas9 and explore the optimal conditions of the system, we need a reporter to indicate the whether the Cas9 functions and the targeted sites are cut off and destructed. So a cell line integrated with EGFP gene is firstly constructed in a similar way. This time the PB-EGFP plasmid (Figure 7), a transposon vector with EGFP gene between the transposon-specific inverted terminal repeat sequences and PiggyBac transposase is co-transfected into cells.
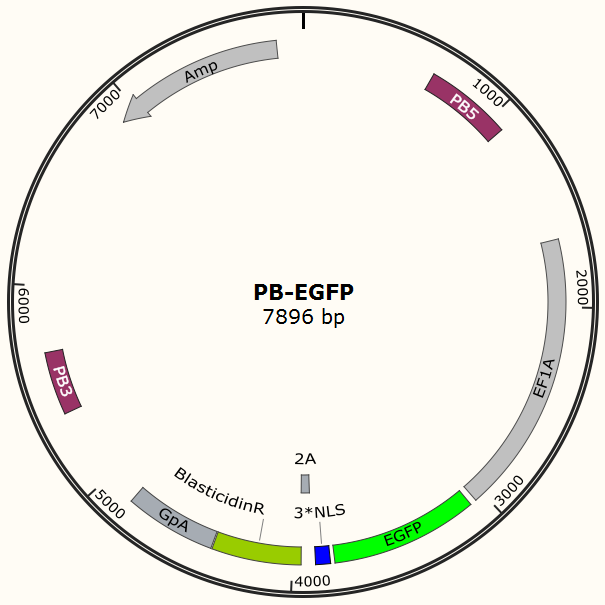
After transfection we treat cell with blasticidin to get a stable cell line expressing EGFP. Then we transiently transfect the cell line with pX330 embedded with gRNA for EGFP and test the efficiency of Cas9-gRNA system.
Deliver plasmids expressing gRNA (guide RNA) by A-B toxin
We choose a non-viral DNA delivery system which is based on modified A-B toxin and yeast GAL4 transcription factor. Yeast transcriptional activator GAL4 protein can specifically recognize plasmids containing UAS sequence. By replacing A subunit of A-B toxin with GAL4, we can get a chimeric fusion protein that can transfer plasmids with UAS into host cells. We construct a plasmid which contains UAS sequence and can transcribe gRNA in human cells, and use it as our gRNA expression vector in human cells.
DNA delivery shuttle design
The chimeric fusion protein mainly comprises 3 parts: target cell-specific binding domain, a translocation domain and a nucleic acid binding domain. The target cell-specific binding domain recognizes the EGF (epidermal growth factor) receptors on the cell surface. The translocation domain enhances nucleic acid escape from the cellular vesicle system and thus to augment nucleic acid transfer. The nucleic acid binding domain, which derives from the yeast GAL4 transcription factor, can carry plasmids with UAS (Upstream Activation Sequence) sequences into cells in vivo. Several research groups from Germany to Taiwan (Gaur, Gupta, Goyal, Wels, & Singh, 2002) (Chen et al., 2000) (Fominaya, Uherek, & Wels, 1998) have achieved this goal. We are requesting the plasmid encoding TEG vehicle from a Germany research group (Wels, Winfried 63110 Rodgau (DE)). Figure 8 shows the schematic representation of the TEG fusion gene in the E.coli expression plasmid pWF47-TEG.

Plasmid & experiment design
Before using this delivery system, we need to check the delivery efficiency of this protein system at first. So, firstly, we construct a plasmid that carries a sgRNA for EGFP, two GAL4 sequences which are necessary for the vehicle’s recognition and binding and a mCherry gene sequence followed by a NLS (nucleus leading sequence). Figure 9 demonstrates the plasmid we construct.
The construction is based on the plasmid backbone of Zhang Lab’s pX330. There are two BbsI restriction enzyme sites between U6 strong promoter and U6 terminator in which we can insert sgRNA. Once sgRNA is inserted, the two BbsI sites disappear and the sgRNA can’t be changed anymore. Figure 10 shows how sgRNA replacement is blocked once BbsI sites are used.
Figure 10 Mechanism of gRNA insertion into pX330-derived plasmid backbone CBh is a eukaryotic promoter, followed by mCherry and 3*NLS. NLS makes red fluorescent protein enter into cell nucleus after expression. By means of PCR and molecular cloning, we can change the number of UAS, a sequence of 17bp which is recognized and bound by GAL4 protein. The sgRNA we used in this plasmid is for EGFP, to collaborate with the Cas9 system and EGFP gene that we have integrated into Hela cell line. In this way, we can visualize how many plasmids are transformed into nucleus by detecting the quantity of red-fluorescence-emitting cells. And with the ratio of reduced green fluorescence and the quantity of red fluorescence we can roughly obtain the efficiency of the CRISPR-Cas system we created. And finally, we reconstruct the plasmid but this time with the sgRNA targeting HIV and test its efficacy.
Content
You can use these subtopics to further explain your project:
- Overall project summary
- Project Details
- Materials and Methods
- The Experiments
- Results
- Data analysis
- Conclusions
It's important for teams to describe all the creativity that goes into an iGEM project, along with all the great ideas your team will come up with over the course of your work.
It's also important to clearly describe your achievements so that judges will know what you tried to do and where you succeeded. Please write your project page such that what you achieved is easy to distinguish from what you attempted.