 "
"
Team:BGU Israel/Modeling/scRNA
From 2014.igem.org
(Created page with "{{Team:BGU_Israel/Menu}} <html> <section> <div style="height:12px"> </div> <div style="margin-bottom:10px"> <img src="https://static.igem.org/mediawiki/2014/5/5f/BG...")
Newer edit →
Revision as of 16:19, 13 October 2014


Overview
A python program we wrote in order to simplify the design stage of the scRNA in the mechanism of ‘Intelligent Medication’. Our program constructs the most suitable scRNAs for this mechanism, out of various automated calculations.
Overview
Purpose: Construct the most suitable scRNA, for the following mechanism.

Figure 1 – Need to put content

Figure 2
We wrote a python program which can create the required scRNA, basing on online and offline calculations. The program gets the trigger gene and the target gene as inputs. The output of the program is a list of scRNAs (A•B) and their detection targets (X), that predicted to be the most effective for this mechanism, considering the trigger and target genes.
How It Works
Main Goal:
Finding X, A and B that will make the optimal distinction between OFF and ON states:
- OFF state - minimal production of Dicer substrate B in the absence of detection target X, the presence of mRNA silencing target Y, or the presence of mRNA off-target Z.
- ON state - strong production of B in the presence of short RNA detection target Xs (‘a-b-c’) or full-length mRNA detection target X.
Steps:
- From the target gene (given as an input), find an appropriate sub-sequence to be the silencing target with a length of |y|+|z|, and consequently define y and z.
- For each optional sub-sequence with the length of X (|a|+|b|+|c|), of the trigger gene (given as an input), do:
- Define this sub-sequence as X.
- Split X into three parts, and consequently define a, b and c.
- Define A = z · c* · b* · a*.
- Define B = z · c* · b · c · z* · y*.
- Scoring the complex of A·B (scRNA) and X. **
- Create a list of all the scRNAs (and their mRNA detection target X), sorted by descending score, and set it as the output. The scRNA with the best score is the first.
** - Using an external online service.
Scoring a scRNA A scRNA’s score consists of two components:
- ScoreOFF (OFF state) – the relative concentration of duplex A•B in the absence of X.
- ScoreON (ON state) – the relative concentration of hairpin B in the presence of X. We tried to find a simple scoring function which can describe the suitability of a scRNA in a proper way. There are two simple functions which are candidates for the scoring function, summation and multiplication. We can see it in the following example:

Figure 3
The idea of this score is that we don’t want a situation which a high score component compensate the other component, this is due to the nature of the mechanism (both states are equally important, with interdependency). Therefore, we chose multiplication as the scoring function:
The scRNA’s total score = ScoreOFF * ScoreON
External online services usage:
- InvivoGen - for finding an appropriate sub-sequence for mRNA detection target.
- NUPACK Server - for analyzing secondary structures and finding the concentration of complexes.
User Manual
System Requirements:
- Python 3.x installed.
- Internet connection.
- Free space of a few gigabytes (depends on the trigger gene length).
Running the program:
- The program consist of two python files that can be downloaded from here: [link 1], [link 2].
- Put the two ‘.py’ files (“serverHandlers.py”, “scRNA.py”) in the same directory.
- Create two files in this directory:
- Trigger file (e.g. “trigger.txt”), which consist the trigger gene sequence.
- Target file (e.g. “target.txt”), which consist the target gene sequence.

- Open the command window and run:
> python scRNAalgo.py trigger_file target_file - During the running of the program, it creates a set of all the results from the servers in the directory, for calculation purposes. After the program ends, an appropriate message will appear on the screen and the results will be written into the output file.

Output File (Results):
During the running, the program creates a directory which called “All Results” (if it not exists). In this directory the program will create a sub-directory for every run, with the time and date that make up its name. After the run, this sub-directory will contain all the data that the program has processed of every potential scRNA.
The output file is located in ‘All Results/000000000000 run/000000000000 run - Output.txt’
The output consists all of the scRNAs the program analyzed, in descending order by the total score. A single scRNA will showed like that:
Score: %%%%%%%%
A: %%%%%%%%%
B: %%%%%%%%%
How We Used It
We ran the program on the following sequences, located in the two input files:
Trigger gene sequence - Apolipoprotein3

Target gene sequence - PTP1B

Results:
The beginning of the output file:
Score: 9.999665000418599e-09
A: UGGAGAAAGGUUCGUUAAACCAUCGGUCACCCAGCCCCUGGCCUGCUG
B: UGGAGAAAGGUUCGUUAAACCAGGCUGGGUGACCGAUGGUUUAACGAACCUUUCUCCAUC
X: CAGCAGGCCAGGGGCUGGGUGACCGAUGG (215 - 243)
Score: 9.999613001825999e-09
A:UGGAGAAAGGUUCGUUAAAUGAGGUCUCAGGCAGCCACGGCUGAAGUU
B:UGGAGAAAGGUUCGUUAAAUGAGGCUGCCUGAGACCUCAUUUAACGAACCUUUCUCCAUC
X:AACUUCAGCCGUGGCUGCCUGAGACCUCA (325 - 353)
Score: 9.9993870000612e-09
A:UGGAGAAAGGUUCGUUAAAGCUCGCAGGAUGGAUAGGCAGGUGGACUU
B:UGGAGAAAGGUUCGUUAAAGCUCUAUCCAUCCUGCGAGCUUUAACGAACCUUUCUCCAUC
X:AAGUCCACCUGCCUAUCCAUCCUGCGAGC (361 - 389)
The scRNA with the best score (highest) is the first:
A: UGGAGAAAGGUUCGUUAAACCAUCGGUCACCCAGCCCCUGGCCUGCUG
B: UGGAGAAAGGUUCGUUAAACCAGGCUGGGUGACCGAUGGUUUAACGAACCUUUCUCCAUC
This scRNA targets X which starting at the 215th basis of the target gene up to the 243th:
X: CAGCAGGCCAGGGGCUGGGUGACCGAUGG (215 - 243)
Results Analysis
Analysis of the chosen scRNA
We chose the first scRNA, and verified the results by putting A and B together in an identical concentration, with and without the trigger gene.
OFF state (without trigger)
No concentration of B (0%) - this is a very good OFF state.
The duplex A·B is the only complex (100%) that formed from A and B:

Figure 3
ON state (with trigger)
There is a concentration of 50% of B and 50% of X·A. That is to say, all of the B forms will transform into hairpins, and will look like in the figure below:
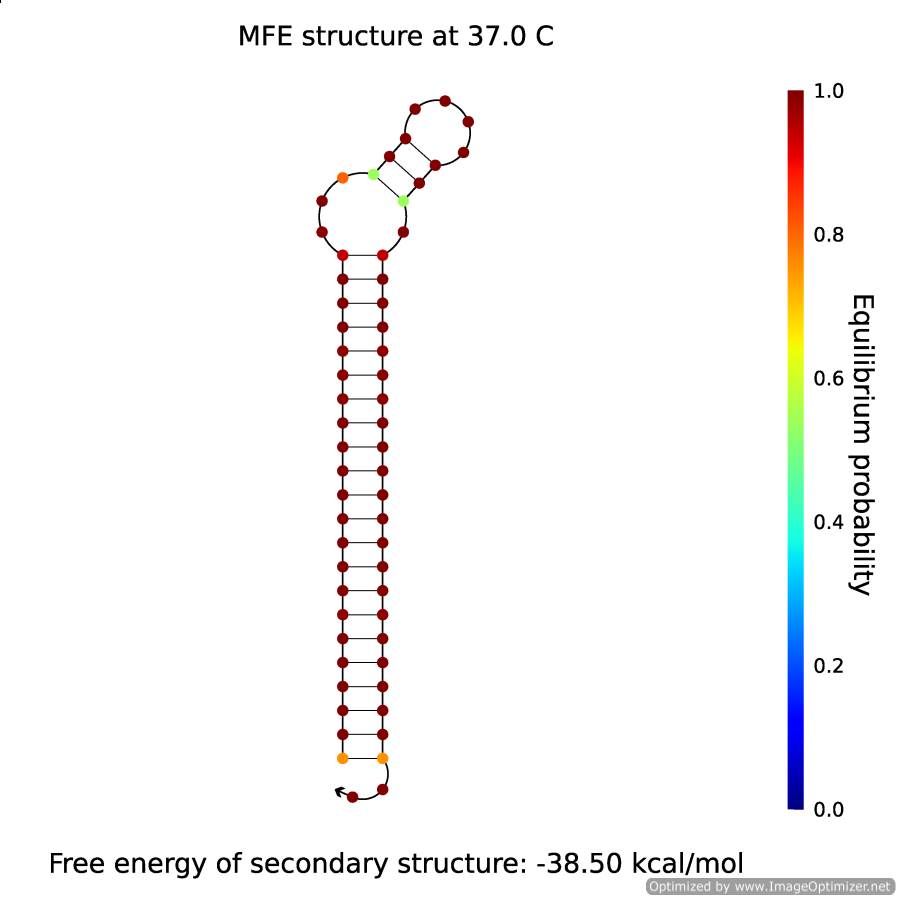
Figure 4
The A which has separated from B, assembles, along with X, the complex X·A:
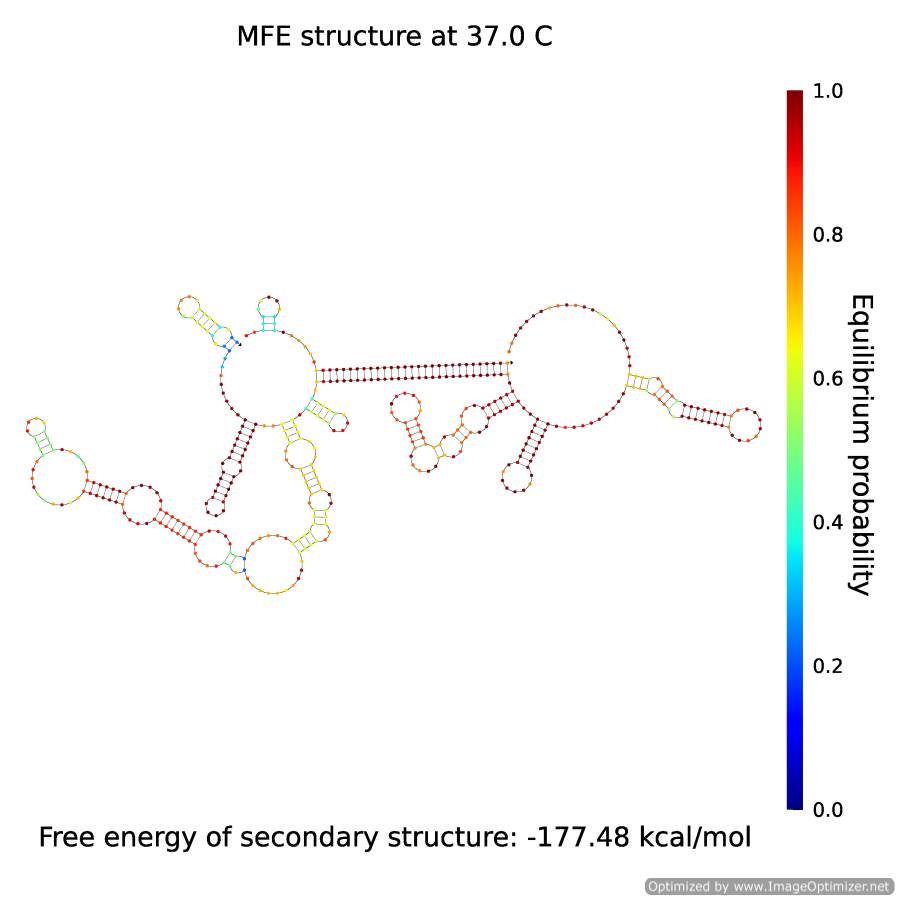
Figure 5
Conclusions:
We used the program we wrote in the design and test of our scRNA mechanism. It helped us to choose the scRNA we wanted to test in our lab, among a very large number of potential scRNAs, and is also even spared us the manual analysis of a single scRNA. We offer the code to of the program as open source, from the notion that it can help anyone who want to design the same mechanism we used, for other purposes. Of course the program is adjustable to the needs of any objective, with a different trigger gene and a different target gene.