From 2014.igem.org
(Difference between revisions)
|
|
| Line 45: |
Line 45: |
| | | | | | |
| | {|style="background:#FFFFFF; margin:0.1em" | | {|style="background:#FFFFFF; margin:0.1em" |
| - | !align="center"|[[File:New ひと.jpg|link=Team:KAIT_Japan/Human_Practice]] | + | !align="center"|[[File:New new ひと.jpg|link=Team:KAIT_Japan/Human_Practice]] |
| | [[Team:KAIT_Japan/Human_Practice|Human Practice]] | | [[Team:KAIT_Japan/Human_Practice|Human Practice]] |
| | |} | | |} |
Revision as of 05:52, 12 September 2014

|
|
|
|
|

Protocol
|
|
|
|
|
|
|
|
Protocol
1:miniprep
- ・We took plasmid out of Escherichia coli which have a gene of IL-10 α and IL-10 β and STAT3 in miniprep to use it by a following experiment (the Escherichia coli which I really used in an experiment of 2013).
DNA of HlyA and the GFP are IGEM 2014 kit plate1 21G and IGEM 2014 kit plate 13L, so we didn't miniplep
- 1) We cultured bacterial strain with the LB medium which I added ampicillin to so that density becomes 100ug/ml overnight.(We made a nutrient medium of around 5 ml in 50 ml falcons)(Against 5 ml of nutrient mediums, Amp used 5ul)
- 2) Aliquot 1ml culture into a 1.5 ml microcentrifuge tube,and Made it spin at 10000rpm (4℃) for 1 min to harvest the bacteria.
- 3) Removed supernatant and performed 2)operation again, Removed supernatant .
- 4) Resuspended bacterial pellet by complete vortexing in 100ul SolutionⅠ{D-glucose:9g(50mM),1M Tris-HCl(pH 8.0):25ml(25mM),0.5M EDTA:20ml(10mM),H2O:955ml /1L}.
- 5) Inverted bacterial pellet by complete fall mixtureing in 200ul SolutionⅡ{NaOH:8g,SDS:10g[1%(w/v)],H2O:960ml /1L},and confirmed that it became transparent.
- 6) Cooled for three minutes in ice.
- 7) Inverted bacterial pellet by complete fall mixtureing in 150ul SolutionⅢ{CH3COOH:294.5g(3M),CH3COOH:120ml(2M),H2O:diluting in measuring cylinder to 1L total},and confirmed that it became Cloudiness.
- 8) Cooled for 3 minutes in ice.
- 9) Harvested the DNA by spinning at 10000rpm (4℃) for 10 min.
- 10) Gathered only supernatant and moved it in a new microcentrifuge tube.
- 11) Added 0.8ul Rnase(10mg/ml) and incubate the solution(37℃,1min)
- 12) Added 200ul phenol:chloroform(1:1) and inverted
- 13) Harvested by spinning at 10000rpm (4℃) for 5 min.
- 14) Removed only supernatant and moved it in a new microcentrifuge tube, after that tapped in 200ul chloroform.
- 15) Harvested by spinning at 10000rpm (4℃) for 1 min.
- 16) Moved its supernatant to a new microcentrifuge tube and add 15ul 3M CH3COONa.(Don't gather underlayer)(The ratio of the 3M sodium acetate and supernatant is made to be 10:1)
- 17) Added 400ul 100%CH3CH2OH and made it stirred well.
- 18) Harvested by spinning at 10000rpm (4℃) for 20 min.
- 19) Removed only supernatant and added 400ul 70%CH3CH2OH(pour a liquid from the other side for white thing not to drain a white.[white thing is plasmid])
- 20) Harvested by spinning at 10000rpm (4℃) for 20 min.
- 21) Removed only supernatant and opened the cover of the tube for 10min to dry CH3CH2OH.
- 22) Added 50ul TE to dissolve DNA
- 23) We stored low temperature
2:PCR
- ・We performed PCR to confirm whether DNA which I got from miniprep and IGEM kit really increased
- 1)We diluted the primer.(D2W:primer=4:1)
- 2)Made PCR preparation liquid {buffer×10:2.5ul,dNTP:2ul,PrimerF:1ul,PrimerR:1ul,Taq Polumerase:0.1ul,D2W:17.39ul /1 microcentrifuge tube(0.2ml)}
- 3)Added sample(there are Plasmid made with miniprep)to the microcentrifuge tube and do PCR(The PCR conditions are as follows).
- Figure①:PCR condition of STAT3 ,IL-10a and IL-10b

- [②→③→④]:repeated 35~40 cycles
- Figure②:PCR condition of GFP and HlyA
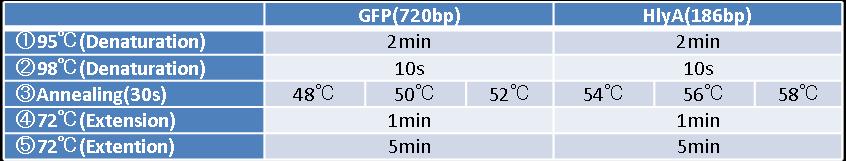
- [②→③→④]:repeated 40 cycles
- 4)We did Electrophoresis to confirm Objective band.
3:Restriction enzyme processing
- ・We did restriction enzyme processing to plasmid to incorporate GFP+HlyA in a plasmid vector.
- Figure③:Used Restriction enzyme
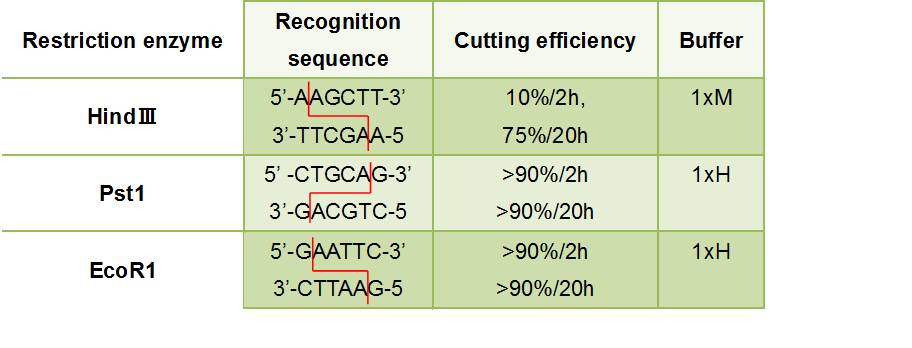
- HlyA・・・We removed the stop codon
- F chain ・・・This has EcoR1 recognition sequence.We added CG and A to this to prevent flame out.
- ・5'-CGGAATTCATTAGCCTATGGAAGTCAGGG-3'
- ((Tm before adding a restriction enzyme site =60℃
- ((GC before adding a restriction enzyme site =50.0%
- R chain ・・・This has HindⅢ recognition sequence.We added CCC to this.
- ・5'-CCCAAGCTTTGCTGATGTGGTCAGGGTTA-3'
- ((Tm before adding a restriction enzyme site =60℃
- ((GC before adding a restriction enzyme site =50.0%
- GFP
- F chain ・・・This has HindⅢ recognition sequence.We added CCC and A to this to prevent flame out.
- ・5'-CCCAAGCTTAATGCGTAAAGGAGAAGAACT-3'
- ((Tm before adding a restriction enzyme site =56℃
- ((GC before adding a restriction enzyme site =40.0%
- R chain ・・・This has Pst1 recognition sequence.We added AA to this.
- ・5'-AACTGCAGTTATTATTTGTATAGTTCATCC-3'
- ((Tm before adding a restriction enzyme site =54℃
- ((GC before adding a restriction enzyme site =22.7%
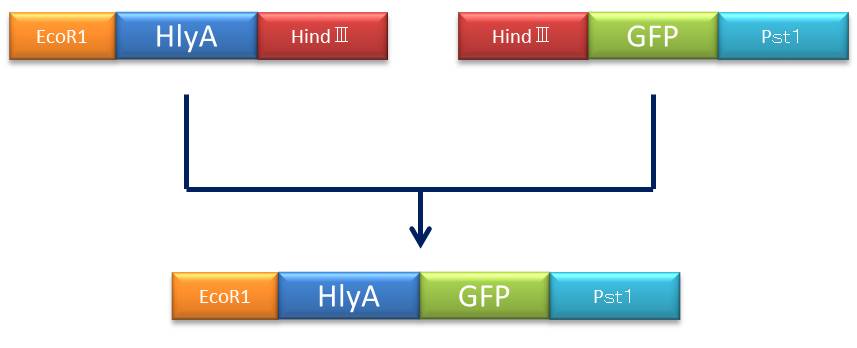
- Figure④:Rough plan
- 1)
- 2)
- 3)
- 4)
4:Transformation
- ・We did restriction enzyme processing to plasmid to incorporate GFP+HlyA in a plasmid vector.And we incorporated GFP+HlyA in plasmid by ligation processing
- 1)We put 1μℓeach plasmid into 3 microtubes.(used plasmid are pCold vectorⅠ(4407bp),pCold vectorⅡ(4392bp) and pSBIC3)
- 2)Put 1ul PstⅠand 1ul EcoRⅠinto each microtube.
- 3)Put 1ul ×10 H buffer into each microtube.
- 4)Up to 10ul with (D2W.
- 5)Did heat block(37℃) about 2hour.
- 6)Prepared 3 new microtubes.and added 10ul GFP+HlyA into them.
- 7)
|
 "
"