 "
"
Team:SUSTC-Shenzhen/Notebook/Biobricks Characterization
From 2014.igem.org
Zhangysh1995 (Talk | contribs) (→9.14) |
Zhangysh1995 (Talk | contribs) |
||
| (9 intermediate revisions not shown) | |||
| Line 10: | Line 10: | ||
='''Scheme'''= | ='''Scheme'''= | ||
| - | At first, we want to characterize plasmid assembled by 3 promoters, 3 RBSs, and 4 chromoprotein (36). Because time limits, we choose 2 promoter, 2 RBSs and 4 chromoprotein (16). | + | Team Uppsala 2012 chromoprotein attracts many interests because its more convenient than fluorescent protein to be use as a reporter. However, few characterized data can be found for these proteins. SUSTC-Shenzhen this year want to tackle with theses problems. At first, we want to characterize plasmid assembled by 3 promoters, 3 RBSs, and 4 chromoprotein (36). Because time limits, we choose 2 promoter, 2 RBSs and 4 chromoprotein (16). However, we found construction with E1010 is very hard because the promoter we used (J23100 & J23106) can express chromoprotein E1010, which makes it difficult and unnecessary to distinguish between these proteins. So we abandoned BBa_E1010 and do experiments on other chromoproteins. |
| + | |||
='''Results'''= | ='''Results'''= | ||
| Line 66: | Line 67: | ||
=='''9.8'''== | =='''9.8'''== | ||
| - | Amplification of biobricks we would use. Our protocol is based on the iGEM | + | Amplification of biobricks we would use. Our protocol is based on the iGEM Protocols:<br> |
| - | Protocols:<br> | + | |
#Punch a hole into the corresponding hole in the hole in 2014 kit plate by a pipette tip. Adding 10ul ddH20 to the hole and pipette up and down for several times. Let sit for 7 min to make sure the dried DNA is fully resuspended. | #Punch a hole into the corresponding hole in the hole in 2014 kit plate by a pipette tip. Adding 10ul ddH20 to the hole and pipette up and down for several times. Let sit for 7 min to make sure the dried DNA is fully resuspended. | ||
#Transform 3ul DNA into E. coli DH5α competent cell. Ice incubate for 30min. | #Transform 3ul DNA into E. coli DH5α competent cell. Ice incubate for 30min. | ||
| Line 171: | Line 171: | ||
No colony formed again. We thought the most likely reason is our competent cells. So we try to repeat the experiments again with the competent cells of our teacher’s. | No colony formed again. We thought the most likely reason is our competent cells. So we try to repeat the experiments again with the competent cells of our teacher’s. | ||
| - | =='''9.26~9.28''' | + | =='''9.26~9.28'''== |
Fortunately, 8 plates we incubate grew out many colonies, and we picked up single colonies from each plate to amplify and extract the plasmids. Then we did enzyme digestion and gel electrophoresis to verify the results. | Fortunately, 8 plates we incubate grew out many colonies, and we picked up single colonies from each plate to amplify and extract the plasmids. Then we did enzyme digestion and gel electrophoresis to verify the results. | ||
| Line 177: | Line 177: | ||
From the result we could see that all plasmids except for B31+K11 are constructed successfully. | From the result we could see that all plasmids except for B31+K11 are constructed successfully. | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
=='''9.29'''== | =='''9.29'''== | ||
| Line 1,480: | Line 1,421: | ||
All 8 biobricks were transformed into BL21 and spread plates. We take pictures for several hours. And single colonies are isolated into LB broth for testing under spectrometer. | All 8 biobricks were transformed into BL21 and spread plates. We take pictures for several hours. And single colonies are isolated into LB broth for testing under spectrometer. | ||
| + | '''See details on page of each parts!''' | ||
='''References'''= | ='''References'''= | ||
| - | #[http://www.tiangen.com/en/?productShow/t1/4/id/32.html | + | #[http://www.tiangen.com/en/?productShow/t1/4/id/32.html TIANprep Mini Plasmid Kit] |
| - | #[http://www.tiangen.com/en/?productShow/t1/4/id/41.html | + | #[http://www.tiangen.com/en/?productShow/t1/4/id/41.html TIANprep Midi Purification Kit] |
| - | #[https://www.neb.com/products/E0546-BioBrick-Assembly-Kit | + | #[http://www.tiangen.com/en/?productShow/t1/4/id/40.html Universal DNA Purification Kit(DP214)] |
| + | #[https://www.neb.com/products/E0546-BioBrick-Assembly-Kit NEB Biobricks® Assembly Kit] | ||
Latest revision as of 23:39, 17 October 2014
Notebook
Biobricks Characterization
Contents
|
Scheme
Team Uppsala 2012 chromoprotein attracts many interests because its more convenient than fluorescent protein to be use as a reporter. However, few characterized data can be found for these proteins. SUSTC-Shenzhen this year want to tackle with theses problems. At first, we want to characterize plasmid assembled by 3 promoters, 3 RBSs, and 4 chromoprotein (36). Because time limits, we choose 2 promoter, 2 RBSs and 4 chromoprotein (16). However, we found construction with E1010 is very hard because the promoter we used (J23100 & J23106) can express chromoprotein E1010, which makes it difficult and unnecessary to distinguish between these proteins. So we abandoned BBa_E1010 and do experiments on other chromoproteins.
Results
We successfully constructed 8 parts, and they all are characterized. And 6 parts were sent to Registry of Standard Biological Parts. See them | HERE.

Procedures
- Amplification of Biobricks
- Add RBS
- Add promoter
- Add terminator

Plasmid Construction
ALL ABBREVIATIONS USED:
| Parts name | Abbreviations | Parts name | Abbreviations |
|---|---|---|---|
| BBa_J23100 | J00 | BBa_E1010 | E10 |
| BBa_J23106 | J06 | BBa_K592009 | K09 |
| BBa_B0031 | B31 | BBa_K592011 | K11 |
| BBa_B0034 | B34 | BBa_K1033916 | K916 |
| BBa_B0015 | B15 | BBa_I20260 | None |
| BBa_J04450 | None |
9.8
Amplification of biobricks we would use. Our protocol is based on the iGEM Protocols:
- Punch a hole into the corresponding hole in the hole in 2014 kit plate by a pipette tip. Adding 10ul ddH20 to the hole and pipette up and down for several times. Let sit for 7 min to make sure the dried DNA is fully resuspended.
- Transform 3ul DNA into E. coli DH5α competent cell. Ice incubate for 30min.
- 42℃ heat shock for 90s, then ice incubate for 5min.
- Adding 200ul SOC medium and shake at 200rpm for 50min.
- Centrifuge 12000rpm for 2min. Discard 200ul supernatant and resuspend the cell pellet in the rest 50ul medium. Then spread the plate with all 50ul broth.
- 37℃ incubate overnight. (19:27)
9.9
We observed the plate at 14:00 and found the bacteria on several plates were growing much faster than on the other plates. We then put the plates which has grown out colonies into the refrigerator in case of overgrowth and continued to incubate the rest plates at 37℃. At 19:00, we found that some plates in the incubator had grown out several colonies.
9.10
At 0:20, we picked up one colony for each plate and used shake incubator. B0015, B0030, K592011 and J04450 didn’t grown out. We redid the transformation and spreading the plate in the day time.
We extracted plasmids (16:00), do the enzyme digestion (21:00) and do gel electrophoresis (23:50).
Gel electrophoresis results:
 From the gel result, we could see that all plasmids were successfully double digested, and the plasmid size was correct.
From the gel result, we could see that all plasmids were successfully double digested, and the plasmid size was correct.
9.11
We picked up the colonies of B0015, B0030, K592011 and J04450 at 19:00 and shake in the incubator overnight.
9.12
We extracted the plasmid (10:00) and do double enzyme digestion (18:10~23:10). Then we did gel electrophoresis to verify the results (23:49).
 From the result we can conclude that all plasmids have been successfully amplified.
From the result we can conclude that all plasmids have been successfully amplified.
9.14
First we would link RBS with Chromoprotein by 3A assembly.
Enzyme digestion:
For RBS (B0031, B0034), using EcoRI-HF and SpeI to cut. For chromoprotein (E1010, K1033916, K592009, K592011), using XbaI and PstI to cut. For the plasmid backbone (I20260, Kana resistance), using EcoRI-HF and PstI.
| Plasmid | 500ng |
|---|---|
| Enzyme I | 1ul |
| Enzyme II | 1ul |
| 10X NEBuffer
2.1 | 5ul |
| ddH20 | to 50ul |
Enzyme digestion: 15:27~16:16
80℃ inactivation: 16:16~16:37
Ligation: 17:15~18:00 (room temperature)
Transformation: Using 5ul ligation product and 50ul competent cell. Heat shock 90s and revive for 2h. Centrifuge and concentrate the broth before spreading the plate (20:00~23:00).
9.15~9.17
The plate grow out with a lot of false colonies (with green fluorescence due to GFP in I20260). We doubt that it was due to the enzyme digestion efficiency and strong self ligation, so we tried to do enzyme digestion and ligation again with the linearized plasmid provided by Parts Registry in 2014 kit. We also recalculated the molar ratio of RBS, chromoprotein and plasmid backbone and try to make it into 10:3:1.
Enzyme digestion (37℃ overnight):
| Plasmid | 20ng |
|---|---|
| Enzyme I | 0.2ul |
| Enzyme II | 0.2ul |
| 10X NEBuffer 2.1 | 0.5ul |
| ddH20 | to 10ul |
Ligation reaction:
| T4 DNA ligase | 0.5ul |
|---|---|
| pSB1K3 linearized | 1ul |
| RBS (10ng/ul) | 6ul |
| Chromo (10ng/ul) | 1.5ul |
| 10 X T4 DNA ligase buffer | 1ul |
| Total | 10ul |
Then we did the transformation as the normal protocol.
9.18~9.22
We did the plasmid backbone recovery and competent cell efficiency test, but we failed. We also picked up several colonies on the first ligation and transformation plate and incubate it in LB broth medium, but we also failed. We thought it’s due to the low efficiency of 3A assembly, so we changed our plan to traditional assembly: cut and recover chromoprotein from digested system and ligate it into RBS backbone.
For the molar ration, most chromoprotein is 700bp long and the RBS backbone is 2100bp long. If we want the molar ratio of chromoprotein:RBS backbone = 3:1, we should add exact same mass of chromoprotein and RBS backbone.
9.23~9.25
We failed again
No colony formed again. We thought the most likely reason is our competent cells. So we try to repeat the experiments again with the competent cells of our teacher’s.
9.26~9.28
Fortunately, 8 plates we incubate grew out many colonies, and we picked up single colonies from each plate to amplify and extract the plasmids. Then we did enzyme digestion and gel electrophoresis to verify the results.
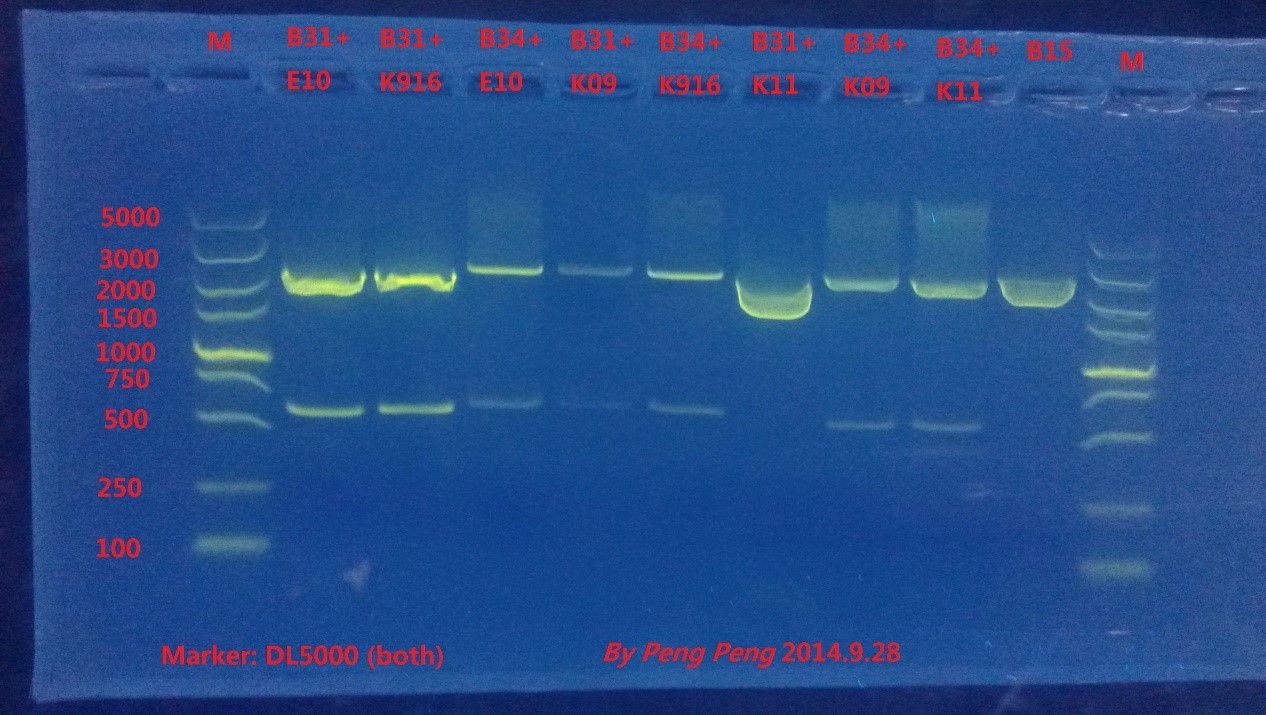
From the result we could see that all plasmids except for B31+K11 are constructed successfully.
9.29
After RBS added, all seven plasmid were cut and ligated with two promoter, J23101 and J23106 respectively.
Enzyme digestion
For plan A
| J00 | J06 | B31E10 | B31K916 | B31K09 | B34E10 | B34K916 | B34 K09 | B34K11 | |
|---|---|---|---|---|---|---|---|---|---|
| EcoRI-HF(μL) | 1 | ||||||||
| XbaI(μL) | 1 | ||||||||
| PstI | 1 | ||||||||
| EcoRV-HF | 1 | ||||||||
| NcoI | 1 | 1 | |||||||
| Linearized
Backbone(μL) | 1 | ||||||||
| DNA(μL) | 3 | 4 | 8 | 7 | 4 | 5 | 5 | 5 | 5 |
| 10X NEB Buffer 2.1(μL) | 5 | ||||||||
| ddH2O (μL) | 39 | 38 | 34 | 35 | 38 | 37 | 37 | 37 | 37 |
| Total(μL) | 40 | ||||||||
For plan B
| J00 | J06 | B31
E10 | B31
K916 | B31
K09 | B34
E10 | B34
K916 | B34 K09 | B34
K11 | |
|---|---|---|---|---|---|---|---|---|---|
| EcoRI-HF(μL) | 1 | ||||||||
| XbaI(μL) | 1 | ||||||||
| PstI | 1 | ||||||||
| NcoI | 1 | 1 | 1 | ||||||
| Linearized
Backbone(μL) | 1 | ||||||||
| DNA(μL) | 3 | 4 | 8 | 7 | 4 | 5 | 5 | 5 | 5 |
| 10X NEB
Buffer 2.1(μL) | 5 | ||||||||
| ddH2O (μL) | 39 | 38 | 34 | 35 | 38 | 37 | 37 | 37 | 37 |
| Total(μL) | 40 | ||||||||
DNA Purification
Follow instructions in kit.
Ligation
To complete construction quickly, we use 3A assembly to achieve plasmid with resistant to chloramphenicol (A) and standard assembly with resistant to Ampicillin (B).
Third step ligation - plan A(3A assembly)
| B331E10 | B31K916 | B31K09 | B34E10 | B34K916 | B34K09 | B34K11 | |
|---|---|---|---|---|---|---|---|
| DNA(50μg) | 4.0μL | 4.0μL | 2.0μL | 4.0μL | 2.0μL | 2.0μL | 4.0μL |
| 10x T4 Ligase
Buffer | 2.0μL | ||||||
| T4 Ligase | 1.0μL | ||||||
| ddH2O | 7.0μL | 7.0μL | 9.0μL | 7.0μL | 9.0μL | 9.0μL | 7.0μL |
| J23100(50μg) | 2.0μL | ||||||
| J23106(50μg) | 2.0μL | ||||||
| Backbone(50μg) | 2.0μL | ||||||
| Total | 10μL | ||||||
Third step ligation -planB(Standard Assembly)
| B31
E10 | B31 K916 | B31 K09 | B34
E10 | B34 K916 | B34
K09 | B34 K11 | |
|---|---|---|---|---|---|---|---|
| DNA(μL) | 0.7 | 0.5 | 1.2 | 0.8 | 1.2 | 1.4 | 0.7 |
| J23100(μL) | 1.2 | ||||||
| J23106(μL) | 1.0 | ||||||
| T4 Ligase(μL) | 0.5 | ||||||
| T4 Ligase buffer(μL) | 1 | ||||||
| ddH2O(μL) | 6.6 6.8 | 6.8
7.0 | 6.1
6.3 | 6.5
6.7 | 6.1
6.3 | 5.9
6.1 | 6.6
6.8 |
| Total(μL) | 10 | ||||||
Ligation: In PCR system, 16 to ligate, 65℃ to inactive, and store at 4℃.
Transformation
- Place 7 EP tubes of 100μL DH5α competent cells on ice from -80℃ to melt.
- Transfer 50μL competent cells to 7 new sterilized EP tubes from each tubes in 1.
- Add 10μL of DNA to one EP tube with competent cells respectively.
- Put all EP tubes on ice for 30mins.
- Incubate in water at 42℃ for 90 seconds, then immediately on ice for 2 minutes.
- Add 200μL SOC broth, then put in a shaking incubator for 40 minutes at 37℃ , 220rpm.
- Centrifuge at 4500rpm for 2minutes, dispose 200μL supernatant.
- Resuspend competent cells and spread plates.
Incubate at 37℃.
9.30
4 plates grew single colonies
J00 B34 E10
J00 B34 K91
J06 B31 K09
J06 B34 K09
Religation
| B34 E10 | B31 K09 | B34 K09 | |
|---|---|---|---|
| DNA(50μg) | 0.8μL | ||
| J23106 | 1.0μL | 1.0μL | |
| J23100 | 1.2μL | ||
| Buffer | 1μL | 1μL | 1μL |
| Ligase | 0.5μL | 0.5μL | 0.5μL |
| ddH2O | 6.5μL | 6.3μL | 6.1μL |
| Total | 20μL | ||
Transformation
Isolate three colonies from plates of J06 B32 E10, J06 B31 K916, J06 B34 E10 and J00 B31 K09 to three tubes of 3ml LB broth respectively.
10.1
Plasmid extraction
J06 B31 K916, J00 B34 K916, J06 B34 K916, J06 B34 E10X2, J06 B31 E10 and J06 B31 K916. (Protocols following instructions in kit.) All these seven plasmid were digested with EcoRI, PstI, EcoRI-HF&PstI and go gel electrophoresis tests on 10.2. (Sample list has the same order in former text)
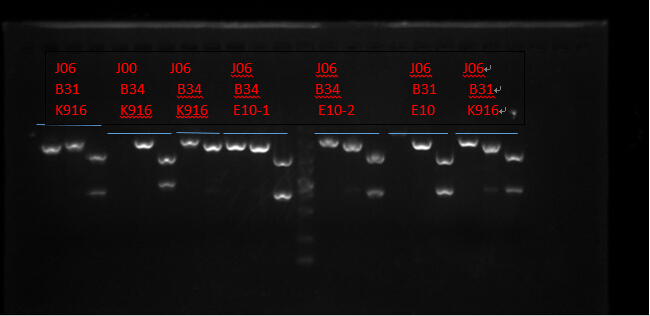
Enzyme Digestion I
| J23100 | J23106 | B31E10 | B31K916 | B31K09 | B34E10 | B34K916 | B34K09 | B34K11 | |
|---|---|---|---|---|---|---|---|---|---|
| DNA(1μg | 3.5μL | 4.1μL | 7.5μL | 6.7μL | 4.4μL | 4.5μL | 4.6μL | 4.6μL | 4.7μL |
| NEB Buffer 2.1 | 5μL | 5μL | 5.0μL | 5.0μL | 5.0μL | 5.0μL | 5.0μL | 5.0μL | 5.0μL |
| XbaI | 0.6μL | ||||||||
| SpeI | 0.6μL | 0.6μL | |||||||
| PstI | 0.6μL | ||||||||
| ddH2O | 40.3μL | 39.7μL | 36.3μL | 37.1μL | 39.4μL | 39.3μL | 39.2μL | 39.2μL | 39.1μL |
| Total | 50μL | ||||||||
Digest Overnight
Enzyme Digestion II
| J06 B31 K916 | J00 B34 K916 | J06 B34 K916 | J06 B34 E10-1 | J06 B34 E10-2 | J06 B31 E10 | J06 B31 K916 | |
|---|---|---|---|---|---|---|---|
| DNA(μL) | 2 | ||||||
| Buffer(μL) | 1 | ||||||
| EcoRI-HF(μL) | 0.3 | ||||||
| PstI(μL) | 0.3 | ||||||
| ddH2O(μL) | 6.4 | ||||||
| Total(μL) | 10 | ||||||
Digest overnight
Enzyme Digestion III
| pSB1C3 RFC | B31K09 | B31 K916 | B31 K09 | B34 E10 | B34 | B34 K11 | B34 | K11 | B31 | J00 | J06 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| XbaI(μL) | 2 | |||||||||||
| PstI(μL) | 2 | |||||||||||
| SpeI(μL) | 2 | |||||||||||
| DNA(μL) | 12 | 15 | 13 | 9 | 9 | 10 | 10 | 10 | 10 | 11 | 7 | 9 |
| 10X NEB Buffer 2.1(μL) | 10 | |||||||||||
| ddH2O(μL) | 74 | 71 | 73 | 77 | 77 | 76 | 76 | 76 | 76 | 75 | 79 | 77 |
| Total | 100 | |||||||||||
Digest overnight.
10.2
Gel electrophoresis
54μL reaction,9μL loading dye
Gel extraction
Ligation
| J00 B31 E10 | J00 B31 K09 | J00 B34 E10 | J00 B34 K09 | J00 B34 K11 | J06 B31 E10 | J06 B31 K09 | J06 B34 E10 | B31 K11 | J00 | J06 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligase(μL) | 0.5 | ||||||||||
| Buffer(μL) | 1 | ||||||||||
| Promoter
Backbone(μL) | 1.3 | 0.9 | 1.3 | ||||||||
| DNA(μL) | 1.2 | 1.5 | 1.6 | 1.6 | 1.6 | 1.2 | 1.5 | 1.6 | 1.4 | ||
| ddH2O(μL) | 6 | 5.7 | 5.6 | 5.6 | 5.6 | 6 | 5.7 | 5.6 | 6.2 | 7.2 | 7.2 |
| Total(μL) | 10 | ||||||||||
Isolate three colonies from plates of J00 B31 K916, J06 B31 K916, J06 B34 K916, J06 B34 K09, J06 B34 K11 to tubes with 3mL LB broth respectively.
10.3
Transformation(10.2)
Enzyme digestion
The five plasmid extracted on 10.1
| J00 B31 K916 | J06 B31 K916 | J06 B34 K916 | J06 B34 K09 | J06 B34 K11 | B0015 | |
|---|---|---|---|---|---|---|
| EcoRI-HF(μL) | 2 | |||||
| SpeI(μL) | 2 | |||||
| DNA(μL) | 9 | 9 | 10 | 9 | 9 | 12 |
| Buffer(μL) | 10 | |||||
| ddH2O(μL) | 77 | 77 | 76 | 77 | 77 | 74 |
| Total(μL) | 100 | |||||
10.4
Gel electrophoresis 120μL (10.3)
Gel extraction
Ligation
Adding terminator and change backbone to pSB1C3.
| J06 B31K916 | J06B34 K916 | J00B31 K916 | J06 B34 K11 | J06 B34 K09 | |
|---|---|---|---|---|---|
| B0015(Gel extraction) (μL) | 1 | ||||
| DNA(μL) | 2 | ||||
| T4 Ligase(μL) | 0.5 | ||||
| T4 Ligase Buffer(μL) | 1 | ||||
| ddH2O(μL) | 5.5 | ||||
| Total(μL) | 10 | ||||
Transformation
Plasmid Extraction B31K11 1~3
Enzyme digestion
B31K11 1~3 were cut with E, P, E&P respectively totally 9 reactions.
Plates (10.2) grow single colonies: J00 B34 E10, J00 B34 K09, J00 B34 K09 isolate single colonies preparing for extraction.
10.5
Plasmid extraction
J00 B34 E10, J00 B34 K09, J00 B34 K11
Gel electrophoresis
J23106 J23100 B31K11
Religation
Ligation products 10.2) grow slowly, which is out of expectation. We decided to redo the ligation.
| J00 B31 E10 | J00 B31 K09 | J00 B34 E10 | J00 B34 K11 | J06 B31 E10 | J06 B31 K09 | J06 B34 E10 | |
|---|---|---|---|---|---|---|---|
| T4 Ligase(μL) | 1 | ||||||
| T4 Ligase Buffer(μL) | 2 | ||||||
| B0015(μL) | 2.6 | ||||||
| DNA(μL) | 2.4 | 3 | 3.2 | 3.2 | 2.4 | 3.0 | 3.2 |
| ddH2O(μL) | 12 | 11.4 | 11.2 | 11.2 | 12 | 11.4 | 11.2 |
| Total(μL) | 20 | ||||||
Transformation
Enzyme digestion
(Preparing for ligation)
| B31K11 | J00 | J06 | |
|---|---|---|---|
| XbaI(μL) | 2 | ||
| PstI(μL) | 2 | ||
| DNA(μL) | 12 | 7 | 9 |
| Buffer(μL) | 10 | ||
| ddH2O(μL) | 74 | 79 | 77 |
| Total(μL) | 100 | ||
Gel electrophoresis
Gel extraction
Ligation
| B31K11 | |
|---|---|
| J00(μL) | 2 |
| J06(μL) | 2 |
| DNA(μL) | 2 |
| Ligase(μL) | 0.5 |
| Buffer(μL) | 1 |
| ddH2O(μL) | 4.5 |
| Total(μL) | 10 |
10.6
(10.5) isolate 3 colonies from the plates.
Ligation
(Adding terminator and changing backbone)
| J00 B34 K11 | J00 B34 K09 | J00 B31 K916 | J06 B31 K916 | J06 B34 K916 | J06 B34 K11 | J06 B34 K09 | |
|---|---|---|---|---|---|---|---|
| B0015(Gel extraction) (μL) | 2 | ||||||
| DNA(μL) | 2 | ||||||
| Ligase(μL) | 0.5 | ||||||
| Buffer(μL) | 1 | ||||||
| ddH2O(μL) | 4.5 | ||||||
| Total(μL) | 10 | ||||||
Transformation
Enzyme digestion I
| J00 B34 E10 | |
|---|---|
| Buffer (μL) | 5 |
| EcoRI-HF(μL) | 1 |
| SpeI(μL) | 1 |
| DNA(μL) | 10 |
| ddH2O(μL) | 33 |
| Total(μL) | 50 |
Enzyme digestion II
| J00 B34 E10 | J00 B34 K09 | J00 B34 K11 | B0015-1 | B0015-2 | B0015-3 | |
|---|---|---|---|---|---|---|
| EcoRI-HF(μL) | 2 | 1 | 2 | |||
| SpeI(μL) | 2 | 1 | ||||
| XbaI(μL) | 2 | |||||
| Buffer(μL) | 10 | 5 | 10 | |||
| DNA(2) (μL) | 13 | 14 | 18 | 5 | 5 | 10 |
| ddH2O(μL) | 73 | 72 | 68 | 39 | 39 | 74 |
| Total(μL) | 100 | 50 | 50 | 100 | ||
We suspected B0015 didn’t work well, and gel electrophoresis was done. B0015 was digested with XbaI, EcoRI-HF, XbaI&EcoRI-HF respectively.

10.7
We checked Part Registry of B0015, and find it’s better to add B0015 behind the coding region instead of inserting protein, RBS and promoter into B0015.
3A assembly
| B0015 | I20260 | J00 B31 K916 | J00 B34 K09 | J00 B34 K11 | J06 B31 K916 | J06 B34 K916 | J06 B34 K09 | |
|---|---|---|---|---|---|---|---|---|
| EcoRI-HF(μL) | 0.5 | 2 | ||||||
| PstI(μL) | 0.5 | 0.5 | 2 | |||||
| 10x NEB Buffer 2.1(μL) | 2 | 5 | ||||||
| DNA(μL) | 11 | 17 | 9 | 14 | 18 | 9 | 10 | 9 |
| ddH2O(μL) | 6 | 4 | 32 | 27 | 23 | 32 | 31 | 32 |
| Total(μL) | 20 | 50 | ||||||
Ligation I
| J06 B31 K916 | J06 B34 K916 | J06 B34 K11 | J06 B34 K09 | J06 B31 K916 | J00 B34 K09 | J00 B34 K11 | |
|---|---|---|---|---|---|---|---|
| B0015(μL) | 1 | ||||||
| I20260(Backbone)(μL) | 1 | ||||||
| T4 Ligase(μL) | 0.5 | ||||||
| T4 Ligase buffer(μL) | 2 | ||||||
| ddH2O | 13 | ||||||
| Total(μL) | 20 | ||||||
Transformation
Ligation II
| J06 B31 E10 | J06 B34 E10 | J06 B34 K11 | J00 B34 E10 | |
|---|---|---|---|---|
| DNA(μL) | 10 | |||
| T4 Ligase(μL) | 0.8 | |||
| T4 Ligase Buffer(μL) | 3 | |||
| ddH2O | 14.2 | |||
| Total(μL) | 30 | |||
Ligation III
| J06 B31 K916 | J00 B31 K916 | J06 B34 K916 | J06 B34 K09 | J00 B34 K11 | J00 B34 K09 | |
|---|---|---|---|---|---|---|
| J04450(Backbone) (μL) | 2.5 | |||||
| T4 Ligase(μL) | 0.4 | |||||
| T4 Ligase buffer(μL) | 014.6 | |||||
| DNA(μL) | 2.5 | |||||
| ddH2O | 14.6 | |||||
| Total(μL) | 20 | |||||
Transformaion
10.8
Plasmid extraction
J00 B34 K11
Isolate three single colonies from plates of J06 B34 K916, J00 B34 K09, J06 B34 K11,J06 B34 K11, J06 B31 K916 (10.6 II).
Ligation
| J00 B34 K916 | J06 B31 K09 | J00 B31 K09 | J00 B31 K11 | J06 B31 K11 | |
|---|---|---|---|---|---|
| J23100(μL) | 2 | 2 | 2 | ||
| J23106(μL) | 2 | 2 | |||
| DNA(μL) | 2 | ||||
| T4 Ligase(μL) | 0.4 | ||||
| T4 Ligase buffer(μL) | 2 | ||||
| ddH2O | 13.6 | ||||
| Total(μL) | 20 | ||||
Transformation with DH5α
10.9
I.Bacteria with 3A assembly plasmid grow so much slowly.
II. Transformation (10.8) was done with DH5α, we have to do again with BL21.
III.Broth culture, plasmid extraction and storage of complete plasmid J00 B34 K09+T, J00 B34K916+T, J06 B34 K09+T, J06 B34 K916+T and J06 B31 K916+T. And extra broth were sent to be sequenced. Unfortunately, EP tubes of J00 B34 K916+T and J06 B31 K916+T were mixed. We recovered broth culture and reinoculated 200μL to 3mL LB Broth, and do extraction again.
V.Plasmid extraction of J06 B31 K916 (without terminator).
Ligation
| J00 B34 K916 | J00 B31 K09 | J00 B31 K11 | J06 B31 K09 | J06 B31 K11 | J06 B34 K11+T | |
|---|---|---|---|---|---|---|
| DNA(μL) | 3 | 3 | 3.5 | 3 | 3.5 | 3 |
| J23100(μL) | 1.5 | 1.5 | 1.5 | |||
| J23106(μL) | 1.5 | 2.5 | ||||
| B0015(μL) | 2.5 | |||||
| T4 Ligase buffer(μL) | 2 | |||||
| T4 Ligase(μL) | 1 | |||||
| ddH2O | 12.5 | 12.5 | 12 | 12.5 | 12 | 11.5 |
| Total(μL) | 20 | |||||
Transformation:First five with BL21, J06 B34 K11+T with DH5α.
VI.Because plasmid constructed with BBa_E1010 is difficult to be differed with J04450, we decided to abandon construction those with BBa_E1010.
10.10
Enzyme digestion
| J00 B31 K916 | B0015 | |
|---|---|---|
| DNA(μL) | 22 | 22 |
| EcoRI-HF(μL) | 2 | 2 |
| SpeI(μL) | 2 | |
| XbaI(μL) | 2 | |
| 10x NEB buffer 2.1(μL) | 5 | 5 |
| ddH2O | 19 | 19 |
| Total(μL) | 50 |
Transformation
10.11
I. Gel electeophoresis
II. Gel extraction
Ligation
| J00 B31 K916 | |
|---|---|
| BOO15(Extraction) | 1.0 |
| DNA | 2 |
| T4 Ligase | 0.5 |
| T4 Ligase
buffer | 2 |
| ddH2O | 13.5 |
| Total | 20 |
Transformation
10.12
I. Plates of (Ampicillin) J06 B31 K916+T, J00 B31 K11+T, J00 B31 K11+T and J00 B31 K09+T grew colonies and single colonies were isolated and broth.
II. Plates of J06 B31 K11, J06 B31 K09, J00 B31 K916 and B0015 grew no colonies.
III. Plates of J06 B34 K11+T (10.9 VI) grew single green colonies one of which was isolated and transfer to broth, and plasmid was extracted at night.
IV. Ligations for J06 B31 K09 and J06 B31 K11 were redid.
Ligation
| J06 B31 K09 | J06 B31 K11 | |
|---|---|---|
| J23106(Backbone) | 1.0μL | |
| DNA(μL) | 2.0μL | |
| T4 Ligase(μL) | 1.5μL | 3.0 |
| T4 Ligase Buffer(μL) | 2μL | 4.0 |
| ddH2O | 13.5 | 10 |
| Total(μL) | 20 | |
10.13
Plasmid extraction
J06 B34 K11+T.
10.14
I.Transformation of J00 B34 K11+T
II.Isolate three single colonies from each plates of J00 B31 K916+T, J00 B34 K11+T, J06 B34 K11+T.
III. Plasmid extraction
IV. Plates of J00 B31 K09 and J00 B31 K11 is not distinct, we decided to abandon these constructions.
Characterization
10.15~10.16
All 8 biobricks were transformed into BL21 and spread plates. We take pictures for several hours. And single colonies are isolated into LB broth for testing under spectrometer.
See details on page of each parts!
References
- [http://www.tiangen.com/en/?productShow/t1/4/id/32.html TIANprep Mini Plasmid Kit]
- [http://www.tiangen.com/en/?productShow/t1/4/id/41.html TIANprep Midi Purification Kit]
- [http://www.tiangen.com/en/?productShow/t1/4/id/40.html Universal DNA Purification Kit(DP214)]
- NEB Biobricks® Assembly Kit