 "
"
Team:SUSTC-Shenzhen/Notebook/CRISPR/mCherry-BbsI-pointmutation
From 2014.igem.org
| (30 intermediate revisions not shown) | |||
| Line 11: | Line 11: | ||
''By Yicong Tao'' | ''By Yicong Tao'' | ||
| - | |||
| - | Plasmid used: pBX-084 PB5-HS4-TRE-mCherrynuc-2A-BlaloxN-GpA-HS4-PB3 (from Wei Huang’s lab). < | + | Plasmid used: pBX-084 PB5-HS4-TRE-mCherrynuc-2A-BlaloxN-GpA-HS4-PB3 (from Wei Huang’s lab). <span style="color: red">Total length is 6843bp.</span> |
| - | Point mutation primer design: | + | Point mutation primer design:<br> |
| + | Forward(5’-3’): cagcccatggttttcttctgcattacggggccg 33nt<br> | ||
| + | Reverse(5’-3’): cggccccgtaatgcagaagaaaaccatgggctg 33nt<br> | ||
| + | The primer pair is intended to eliminate the BbsI site in the mCherry because we need to use BbsI to insert our gRNA into our plasmid. Designed by Agilent Technologies point-mutation primer online design tools. The primers are complement with each other. | ||
| - | + | =Agilent's Pointmutation method= | |
| + | 8.13~8.14 | ||
| + | ==PCR reaction== | ||
| + | (12:06~14:XX 13th Aug, about 2h) | ||
| - | + | '''300ul, 50ul/reaction:''' | |
| - | + | {| class="table" | |
| - | + | ! Component | |
| + | ! Volume | ||
| + | ! Final Concentration | ||
| + | |- | ||
| + | | Q5 master mix 2X | ||
| + | | 150ul | ||
| + | | 1X | ||
| + | |- | ||
| + | | DNA template (251.4ng/ul) | ||
| + | | 1.5ul | ||
| + | | 1.257ng/ul | ||
| + | |- | ||
| + | | Primer F (50uM) | ||
| + | | 3ul | ||
| + | | 500nM | ||
| + | |- | ||
| + | | Primer R (50uM) | ||
| + | | 3ul | ||
| + | | 500nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 142.5ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 300ul | ||
| + | | - | ||
| + | |} | ||
| + | |||
| + | '''PCR reaction conditions:''' | ||
| + | |||
| + | {| class="table" | ||
| + | ! Step | ||
| + | ! Temp (℃) | ||
| + | ! Time (s) | ||
| + | |- | ||
| + | | Initial denaturation | ||
| + | | 98 | ||
| + | | 60 | ||
| + | |- | ||
| + | | 18 cycles | ||
| + | | 98 | ||
| + | | 10 | ||
| + | |- | ||
| + | | | ||
| + | | 58, 60, 61, 62, 63, 65 | ||
| + | | 15 | ||
| + | |- | ||
| + | | | ||
| + | | 72 | ||
| + | | 360 | ||
| + | |- | ||
| + | | Final extension | ||
| + | | 72 | ||
| + | | 120 | ||
| + | |- | ||
| + | | Hold | ||
| + | | 4 | ||
| + | | ∞ | ||
| + | |} | ||
| - | + | ==DpnI digestion== | |
| - | + | (13 Aug 15:05~19:06) | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | |||
Add 1ul DpnI directly to the amplification system | Add 1ul DpnI directly to the amplification system | ||
| + | |||
Digest 4h | Digest 4h | ||
| + | |||
No termination | No termination | ||
| + | |||
Stored at -20℃ | Stored at -20℃ | ||
| - | Nanodrop test after digestion | + | ==Nanodrop test after digestion== |
| - | Anneal Temp (℃) 58 60 61 62 | + | {| class="table" |
| - | Conc (ng/ul) 467.4 465.9 470.4 457.9 | + | ! Anneal Temp (℃) |
| - | 260/280 1. | + | ! 58 |
| - | 260/230 0.73 0.73 0.74 0.73 | + | ! 60 |
| + | ! 61 | ||
| + | ! 62 | ||
| + | |- | ||
| + | | Conc (ng/ul) | ||
| + | | 467.4 | ||
| + | | 465.9 | ||
| + | | 470.4 | ||
| + | | 457.9 | ||
| + | |- | ||
| + | | 260/280 | ||
| + | | 1.8 | ||
| + | | 1.8 | ||
| + | | 1.8 | ||
| + | | 1.81 | ||
| + | |- | ||
| + | | 260/230 | ||
| + | | 0.73 | ||
| + | | 0.73 | ||
| + | | 0.74 | ||
| + | | 0.73 | ||
| + | |} | ||
| + | |||
| + | ==Bacteria transformation== | ||
| + | (2014.8.13 22:08~2014.8.14 14:50) | ||
| + | |||
| + | '''Procedures:''' | ||
| + | |||
| + | #Immediately, place the tubes on ice, allow cell to thaw on ice, 10 min | ||
| + | #Check the cell to see if they have thawed, gently flick the cells 1-2 times to evenly resuspend the cells. | ||
| + | #Divide the competent cells into 6 tubes,50μL each tube, Mark them with 58, 60, 61, 62, 63, 65. Keep the bacteria in ice during the procedure. | ||
| + | #Add 5μL plasmid to each tube, shaking slightly | ||
| + | #Incubate on ice for 30min | ||
| + | #Heat shock, 42°C, 90s. | ||
| + | #Keep on ice, 2 min | ||
| + | #Add 200μl SOC medium to each tube, incubate 37°C, 200rpm, 45min | ||
| + | #Centrifuge, 4000rmp, 5min, RT. Discard most of the medium and reserve 50μL. Resuspend. | ||
| + | #Add 50μL bacteria to amp LB agar plate, distribute. | ||
| + | #Incubate the plates at 37°C, 16 hours. | ||
| + | |||
| + | '''Results:''' | ||
| + | |||
| + | {{SUSTC-Image|wiki/images/e/e1/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-colony.jpg|Bacteria transformation results}} | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
For all 6 groups, no colony forms. | For all 6 groups, no colony forms. | ||
| - | Gel electrophoresis | + | ==Gel electrophoresis== |
| - | Making the agarose gel (1%) | + | '''Making the agarose gel (1%)''' |
| - | Component Volume | + | |
| - | TAE 25ml | + | {| class="table" |
| - | Agarose 0.25g | + | ! Component |
| - | Gene Green 0.5ul | + | ! Volume |
| - | Adding samples | + | |- |
| - | Component Volume | + | | TAE |
| - | PCR product 1ul | + | | 25ml |
| - | 6X loading dye 2ul | + | |- |
| - | TAE 9ul | + | | Agarose |
| - | Total 12ul | + | | 0.25g |
| + | |- | ||
| + | | Gene Green | ||
| + | | 0.5ul | ||
| + | |} | ||
| + | |||
| + | '''Adding samples''' | ||
| + | |||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | |- | ||
| + | | PCR product | ||
| + | | 1ul | ||
| + | |- | ||
| + | | 6X loading dye | ||
| + | | 2ul | ||
| + | |- | ||
| + | | TAE | ||
| + | | 9ul | ||
| + | |- | ||
| + | | Total | ||
| + | | 12ul | ||
| + | |} | ||
| + | |||
DNA marker (Takara DL5000, diluted): 10ul | DNA marker (Takara DL5000, diluted): 10ul | ||
| - | Results: | + | |
| - | + | '''Results:''' | |
| + | |||
| + | {{SUSTC-Image|wiki/images/c/cb/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-afterDpnI-8.13.jpg}} | ||
| + | |||
The result of electrophoresis is not clear, so I redid the gel electrophoresis with the same sample. | The result of electrophoresis is not clear, so I redid the gel electrophoresis with the same sample. | ||
| - | + | ||
| + | {{SUSTC-Image|wiki/images/5/59/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-afterDpnI-8.14.jpg}} | ||
| + | |||
No wanted band (6843bp) is observed. | No wanted band (6843bp) is observed. | ||
Discussion: | Discussion: | ||
| - | + | #PCR failed, so improving PCR conditions is the first choice. Try extending PCR cycles, denaturation time, annealing time and final extension time. | |
| - | + | #Do reaction termination after DpnI enzyme digestion (80℃ 20min) next time because reactive enzyme may binding to the DNA substrate and interfere the transformation efficiency. | |
| - | + | #The concentration of template DNA is also very essential, so using 2-step concentration ladder next time. | |
| + | =Agilent's Pointmutation method II= | ||
8.15 | 8.15 | ||
| - | + | ==PCR== | |
| - | + | (23:35 14th Aug-08:30 15th Aug), about 2h. Next morning, check PCR machine was not at 4℃ hold conditions, and the heating stand was not cold<br> | |
| - | 200ul, 50ul/reaction: | + | '''200ul, 50ul/reaction:''' |
| - | Component Volume Final Concentration | + | {| class="table" |
| - | Q5 master mix 2X 100ul 1X | + | ! Component |
| - | DNA template (251.4ng/ul) 1.0ul (A,B), 2.0ul (C,D) 1.257ng/ul (A,B), 2.514ng/ul (C,D) | + | ! Volume |
| - | Primer F (50uM) 2ul 500nM | + | ! Final Concentration |
| - | Primer R (50uM) 2ul 500nM | + | |- |
| - | ddH20 95ul - | + | | Q5 master mix 2X |
| - | + | | 100ul | |
| - | + | | 1X | |
| - | + | |- | |
| - | + | | DNA template (251.4ng/ul) | |
| - | + | | 1.0ul (A,B), 2.0ul (C,D) | |
| - | + | | 1.257ng/ul (A,B), 2.514ng/ul (C,D) | |
| - | + | |- | |
| - | + | | Primer F (50uM) | |
| - | + | | 2ul | |
| + | | 500nM | ||
| + | |- | ||
| + | | Primer R (50uM) | ||
| + | | 2ul | ||
| + | | 500nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 95ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 200ul | ||
| + | | - | ||
| + | |} | ||
| - | + | '''PCR reaction conditions:''' | |
| - | + | {| class="table" | |
| - | + | ! Step | |
| - | + | ! Temp (℃) | |
| - | + | ! Time (s) | |
| + | |- | ||
| + | | Initial denaturation | ||
| + | | 98 | ||
| + | | 60 | ||
| + | |- | ||
| + | | rowspan="3" | 25 cycles | ||
| + | | 98 | ||
| + | | 20 | ||
| + | |- | ||
| + | | 63 | ||
| + | | 30 | ||
| + | |- | ||
| + | | 72 | ||
| + | | 420 | ||
| + | |- | ||
| + | | Final extension | ||
| + | | 72 | ||
| + | | 600 | ||
| + | |- | ||
| + | | Hold | ||
| + | | 4 | ||
| + | | ∞ | ||
| + | |} | ||
| - | + | ==Nanodrop test== | |
| - | + | {| class="table" | |
| + | ! Anneal Temp (℃) | ||
| + | ! A | ||
| + | ! B | ||
| + | ! C | ||
| + | ! D | ||
| + | |- | ||
| + | | Conc (ng/ul) | ||
| + | | 474.2 | ||
| + | | 492.0 | ||
| + | | 466.7 | ||
| + | | 445.9 | ||
| + | |- | ||
| + | | 260/280 | ||
| + | | 1.82 | ||
| + | | 1.81 | ||
| + | | 1.81 | ||
| + | | 1.81 | ||
| + | |- | ||
| + | | 260/230 | ||
| + | | 0.72 | ||
| + | | 0.73 | ||
| + | | 0.72 | ||
| + | | 0.70 | ||
| + | |} | ||
| - | + | ==Gel electrophoresis== | |
| + | '''Making the agarose gel (1%)''' | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | |- | ||
| + | | TAE | ||
| + | | 25ml | ||
| + | |- | ||
| + | | Agarose | ||
| + | | 0.25g | ||
| + | |- | ||
| + | | Gene Green | ||
| + | | 0.5ul | ||
| + | |} | ||
| + | '''Adding samples''' | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | |- | ||
| + | | PCR product | ||
| + | | 5ul | ||
| + | |- | ||
| + | | 6X loading dye | ||
| + | | 2ul | ||
| + | |- | ||
| + | | TAE | ||
| + | | 5ul | ||
| + | |- | ||
| + | | Total | ||
| + | | 12ul | ||
| + | |} | ||
| + | DNA marker (Takara DL5000, diluted): 10ul | ||
| - | 2-pair primers PCR | + | '''Results:''' |
| + | {{SUSTC-Image|wiki/images/e/e7/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-2nd-PCR.jpg}} | ||
| + | |||
| + | '''Analysis:'''<br> | ||
| + | The band was in 2000bp location, which is far away from the original plasmid length (6800bp), which meant PCR didn’t got complete plasmid.<br> The possible reason might be: | ||
| + | #The primer forms dimers which strongly influence the efficiency of PCR (The smallest band is very light). | ||
| + | #DNA is a double helix, which means the plasmid double strand can also anneal with each other. The elongation time was very long, which left enough time for the plasmid double strand to anneal with each other and prohibit the PCR process. | ||
| + | '''Solution:''' | ||
| + | #Redesign the primer so that the primer pairs are not totally complementary with each other. Put the point mutation point near the 5’ end. | ||
| + | #Use other PCR methods. | ||
| + | |||
| + | <span style="color: red">'''So I decided to change the PCR method.'''</span> | ||
| + | |||
| + | =2-pair primers PCR= | ||
I had two pairs of primers: one is for mCherry (called A), another is for point mutation (called B). I then used the forward primer of A to pair with the reverse primer of B, so did the forward primer of B with the reverse primer of A. Thus, I would get 2 short segments which had a small complementary region. Then, I added the two segments together and let them do PCR using each other as the template and primer in order to get the final whole length PCR product. | I had two pairs of primers: one is for mCherry (called A), another is for point mutation (called B). I then used the forward primer of A to pair with the reverse primer of B, so did the forward primer of B with the reverse primer of A. Thus, I would get 2 short segments which had a small complementary region. Then, I added the two segments together and let them do PCR using each other as the template and primer in order to get the final whole length PCR product. | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | + | '''mCherry primer design:'''<br> | |
| - | + | Forward(5’-3’): TCGTTTAGTGAACCGTCAG 19nt<br> | |
| - | Component Volume | + | Reverse(5’-3’): cgaggaattcctattaTTCCTCTGCCCTCACTAG 34nt<br> |
| - | + | ==PCR== | |
| - | + | '''50ul, 25ul/reaction:'''<br> | |
| - | + | ''Reaction 1:'' | |
| - | + | {| class="table" | |
| - | Component Volume | + | ! Component |
| - | + | ! Volume | |
| - | + | ! Final Concentration | |
| - | + | |- | |
| - | + | | Q5 master mix 2X | |
| - | + | | 25ul | |
| - | + | | 1X | |
| - | + | |- | |
| - | + | | pBX084 diluted (25.14ng/ul) | |
| - | + | | 2.0ul | |
| - | + | | 1.257ng/ul | |
| - | + | |- | |
| - | + | | mCherry Primer F (50uM) | |
| + | | 0.5ul | ||
| + | | 500nM | ||
| + | |- | ||
| + | | Pointmutation Primer R (50uM) | ||
| + | | 0.5ul | ||
| + | | 500nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 22ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 50ul | ||
| + | | - | ||
| + | |} | ||
| + | ''Reaction 2:'' | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | ! Final Concentration | ||
| + | |- | ||
| + | | Q5 master mix 2X | ||
| + | | 25ul | ||
| + | | 1X | ||
| + | |- | ||
| + | | pBX084 diluted (25.14ng/ul) | ||
| + | | 2.0ul | ||
| + | | 1.257ng/ul | ||
| + | |- | ||
| + | | mCherry Primer R (50uM) | ||
| + | | 0.5ul | ||
| + | | 500nM | ||
| + | |- | ||
| + | | Pointmutation Primer F (50uM) | ||
| + | | 0.5ul | ||
| + | | 500nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 22ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 50ul | ||
| + | | - | ||
| + | |} | ||
| + | '''PCR reaction conditions:''' | ||
| + | {| class="table" | ||
| + | ! Step | ||
| + | ! Temp (℃) | ||
| + | ! Time (s) | ||
| + | |- | ||
| + | | Initial denaturation | ||
| + | | 98 | ||
| + | | 60 | ||
| + | |- | ||
| + | | rowspan="3" | 25 cycles | ||
| + | | 98 | ||
| + | | 20 | ||
| + | |- | ||
| + | | 61 | ||
| + | | 15 | ||
| + | |- | ||
| + | | 72 | ||
| + | | 20 | ||
| + | |- | ||
| + | | Final extension | ||
| + | | 72 | ||
| + | | 300 | ||
| + | |- | ||
| + | | Hold | ||
| + | | 4 | ||
| + | | ∞ | ||
| + | |} | ||
| - | + | ==Gel electrophoresis== | |
| - | PCR | + | '''Making the agarose gel (1%)''' |
| - | + | {| class="table" | |
| - | + | ! Component | |
| - | + | ! Volume | |
| - | + | |- | |
| - | + | | TAE | |
| - | + | | 25ml | |
| - | + | |- | |
| + | | Agarose | ||
| + | | 0.25g | ||
| + | |- | ||
| + | | Gene Green | ||
| + | | 0.5ul | ||
| + | |} | ||
| + | '''Adding samples''' | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | |- | ||
| + | | PCR product | ||
| + | | 10ul | ||
| + | |- | ||
| + | | 6X loading dye | ||
| + | | 2ul | ||
| + | |- | ||
| + | | Total | ||
| + | | 12ul | ||
| + | |} | ||
| - | + | DNA marker (Takara DL5000, diluted): 30ul<br> | |
| + | {{SUSTC-Image|wiki/images/d/de/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-first-PCR-result.jpg}} | ||
| + | |||
| + | '''Results:'''<br> | ||
| + | A2: reaction 1 (A: mCherry primer F; 2: Pointmutation Primer R)<br> | ||
| + | 1B: reaction 2 (B: mCherry primer R; 1: Pointmutation Primer F)<br> | ||
| - | + | '''Analysis:'''<br> | |
| - | + | The predicting results is 448bp band in A2 and 372bp band in 1B (almost equally separate the mCherry). But the band in A2 was in 800bp and no band is observed in 1B. The possible reasons might be:<br> | |
| - | + | #I added wrong primer pairs: two mCherry primers for A2 and two pointmutation primers for 1B. So that in A2 I got mCherry and in 1B I got primer dimer. | |
| - | + | #The elongation time might be too long. | |
| - | + | ||
| - | + | <span style="color: red">'''So I redid the 2-pari primers PCR with modified PCR reaction conditions.'''</span> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ==PCR II== | |
| - | + | '''Reaction conditions:''' | |
| - | + | {| class="table" | |
| + | ! Step | ||
| + | ! Temp (℃) | ||
| + | ! Time (s) | ||
| + | |- | ||
| + | | Initial denaturation | ||
| + | | 98 | ||
| + | | 60 | ||
| + | |- | ||
| + | | rowspan="3" | 25 cycles | ||
| + | | 98 | ||
| + | | 20 | ||
| + | |- | ||
| + | | 61 | ||
| + | | 15 | ||
| + | |- | ||
| + | | 72 | ||
| + | | 15 | ||
| + | |- | ||
| + | | Final extension | ||
| + | | 72 | ||
| + | | 120 | ||
| + | |- | ||
| + | | Hold | ||
| + | | 4 | ||
| + | | ∞ | ||
| + | |} | ||
| - | Gel electrophoresis: | + | ==Gel electrophoresis:== |
| - | + | {{SUSTC-Image|wiki/images/c/c2/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-second-PCR-result.jpg}} | |
| - | Results: | + | '''Results:'''<br> |
| - | + | No band is observed.<br> | |
| - | Analysis: | + | '''Analysis:'''<br> |
| - | + | #The elongation time might be too short… | |
| - | + | #The annealing time for 1B might be wrong. | |
| - | I | + | =2-pari primers PCR II= |
| + | 8.16 10:25am~11.25am<br> | ||
| + | I set a temperature gradient (45, 48, 51, 54, 57, 60℃) without shortening the elongation time. | ||
| + | ==PCR== | ||
| + | 150ul, 25ul/reaction:<br> | ||
| + | ''Reaction 1:'' | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | ! Final Concentration | ||
| + | |- | ||
| + | | Q5 master mix 2X | ||
| + | | 25ul | ||
| + | | 1X | ||
| + | |- | ||
| + | | pBX084 diluted (25.14ng/ul) | ||
| + | | 0.5ul | ||
| + | | 12.57ng | ||
| + | |- | ||
| + | | Pointmutation Primer F (50uM) | ||
| + | | 1ul | ||
| + | | 1000nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 23ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 49.5ul | ||
| + | | - | ||
| + | |} | ||
| + | ''Reaction 2:'' | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | ! Final Concentration | ||
| + | |- | ||
| + | | Q5 master mix 2X | ||
| + | | 25ul | ||
| + | | 1X | ||
| + | |- | ||
| + | | pBX084 diluted (25.14ng/ul) | ||
| + | | 0.5ul | ||
| + | | 12.57ng | ||
| + | |- | ||
| + | | Pointmutation Primer R (50uM) | ||
| + | | 1ul | ||
| + | | 1000nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 23ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 49.5ul | ||
| + | | - | ||
| + | |} | ||
| + | '''PCR reaction conditions:''' | ||
| + | {| class="table" | ||
| + | ! Step | ||
| + | ! Temp (℃) | ||
| + | ! Time (s) | ||
| + | |- | ||
| + | | Initial denaturation | ||
| + | | 98 | ||
| + | | 60 | ||
| + | |- | ||
| + | | rowspan="3" | 25 cycles | ||
| + | | 98 | ||
| + | | 20 | ||
| + | |- | ||
| + | | 63 | ||
| + | | 15 | ||
| + | |- | ||
| + | | 72 | ||
| + | | 5min | ||
| + | |- | ||
| + | | Final extension | ||
| + | | 72 | ||
| + | | 10min | ||
| + | |- | ||
| + | | Hold | ||
| + | | 4 | ||
| + | | ∞ | ||
| + | |} | ||
| - | + | ==Gel electrophoresis== | |
| - | + | 11:50am ~12:25pm<br> | |
| + | {{SUSTC-Image|wiki/images/7/74/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-2-pair-1.JPG}} | ||
| + | Unfortunately, only in A2 did I observe the band. I doubt the annealing temperature was not high enough because strong primer dimers forms in 1B group. So I redid the 2-pair primers PCR again with even higher temperature gradient (62,63,64,65,66,67℃). | ||
| + | ==Gel electrophoresis II== | ||
| + | {{SUSTC-Image|wiki/images/c/c1/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-2-pair-2.JPG}} | ||
| + | '''Results:'''<br> | ||
| + | Only vague bands were observed in A2. Still no distinct bands observed in 1B.<br> | ||
| + | '''Analysis:'''<br> | ||
| + | #The reverse primer of pointmutation might be very easy to form dimer with the forward primer of mCherry, which led to the failure of PCR. | ||
| + | #The compatibility of the two separate primers might be very bad. | ||
| - | + | <span style="color: red">'''I decided to try another methods.'''</span> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | Double primer PCR | + | =Half-plasmid PCR & Double primer PCR= |
| - | Mix the two pairs of primer together but the density of the pointmutation primer is only 1/10 of the mCherry primer. So the pointmutation primer will be consumed very quickly in the first several rounds. | + | ==Half-plasmid PCR== |
| - | 150ul, 25ul/reaction: | + | First, only add one direction primer so I could get half linear plasmid. After that, I mix the two DNA together to get the complete DNA.<br> |
| - | Component Volume Final Concentration | + | 50ul, 25ul/reaction:<br> |
| - | Q5 master mix 2X 75ul 1X | + | ''Reaction 1''<br> |
| - | pBX084 diluted (25.14ng/ul) 1ul 25.14ng | + | {| class="table" |
| - | mCherry Primer F&R (50uM) 2ul 6666.7nM | + | ! Component |
| - | Pointmutation Primer R (50uM) 0.2ul 666.7nM | + | ! Volume |
| - | ddH20 69.6ul - | + | ! Final Concentration |
| - | Total 150ul - | + | |- |
| - | PCR reaction conditions: | + | | Q5 master mix 2X |
| - | Step Temp (℃) Time (s) | + | | 25ul |
| - | Initial denaturation 98 60 | + | | 1X |
| - | 25 cycles 98 20 | + | |- |
| - | + | | pBX084 diluted (25.14ng/ul) | |
| - | + | | 0.5ul | |
| - | Final extension 72 5min | + | | 12.57ng |
| - | Hold 4 ∞ | + | |- |
| + | | Pointmutation Primer F (50uM) | ||
| + | | 1ul | ||
| + | | 1000nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 23ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 49.5ul | ||
| + | | - | ||
| + | |} | ||
| + | ''Reaction 2''<br> | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | ! Final Concentration | ||
| + | |- | ||
| + | | Q5 master mix 2X | ||
| + | | 25ul | ||
| + | | 1X | ||
| + | |- | ||
| + | | pBX084 diluted (25.14ng/ul) | ||
| + | | 0.5ul | ||
| + | | 12.57ng | ||
| + | |- | ||
| + | | Pointmutation Primer R (50uM) | ||
| + | | 1ul | ||
| + | | 1000nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 23ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 49.5ul | ||
| + | | - | ||
| + | |} | ||
| + | '''PCR reaction conditions:''' | ||
| + | {| class="table" | ||
| + | ! Step | ||
| + | ! Temp (℃) | ||
| + | ! Time (s) | ||
| + | |- | ||
| + | | Initial denaturation | ||
| + | | 98 | ||
| + | | 60 | ||
| + | |- | ||
| + | | rowspan="3" | 25 cycles | ||
| + | | 98 | ||
| + | | 20 | ||
| + | |- | ||
| + | | 63 | ||
| + | | 15 | ||
| + | |- | ||
| + | | 72 | ||
| + | | 5min | ||
| + | |- | ||
| + | | Final extension | ||
| + | | 72 | ||
| + | | 10min | ||
| + | |- | ||
| + | | Hold | ||
| + | | 4 | ||
| + | | ∞ | ||
| + | |} | ||
| + | ==Double primer PCR== | ||
| + | Mix the two pairs of primer together but the density of the pointmutation primer is only 1/10 of the mCherry primer. So the pointmutation primer will be consumed very quickly in the first several rounds.<br> | ||
| + | 150ul, 25ul/reaction:<br> | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | ! Final Concentration | ||
| + | |- | ||
| + | | Q5 master mix 2X | ||
| + | | 75ul | ||
| + | | 1X | ||
| + | |- | ||
| + | | pBX084 diluted (25.14ng/ul) | ||
| + | | 1ul | ||
| + | | 25.14ng | ||
| + | |- | ||
| + | | mCherry Primer F&R (50uM) | ||
| + | | 2ul | ||
| + | | 6666.7nM | ||
| + | |- | ||
| + | | Pointmutation Primer R (50uM) | ||
| + | | 0.2ul | ||
| + | | 666.7nM | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 69.6ul | ||
| + | | - | ||
| + | |- | ||
| + | | Total | ||
| + | | 150ul | ||
| + | | - | ||
| + | |} | ||
| + | '''PCR reaction conditions:''' | ||
| + | {| class="table" | ||
| + | ! Step | ||
| + | ! Temp (℃) | ||
| + | ! Time (s) | ||
| + | |- | ||
| + | | Initial denaturation | ||
| + | | 98 | ||
| + | | 60 | ||
| + | |- | ||
| + | | rowspan="3" | 25 cycles | ||
| + | | 98 | ||
| + | | 20 | ||
| + | |- | ||
| + | | 45,48,52,55,58,61 | ||
| + | | 20 | ||
| + | |- | ||
| + | | 72 | ||
| + | | 20 | ||
| + | |- | ||
| + | | Final extension | ||
| + | | 72 | ||
| + | | 5min | ||
| + | |- | ||
| + | | Hold | ||
| + | | 4 | ||
| + | | ∞ | ||
| + | |} | ||
The two PCR methods proceed simultaneously from 17:00 to 19:30, then reserved in 4℃. | The two PCR methods proceed simultaneously from 17:00 to 19:30, then reserved in 4℃. | ||
| - | 8.17 | + | ==Gel electrophoresis== |
| - | + | 8.17 | |
| - | + | {{SUSTC-Image|wiki/images/f/fc/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-half-plasmid-double-primer-PCR.jpg}} | |
| - | Analysis: | + | '''Analysis:'''<br> |
| - | From Double-primer PCR we got 2 (or 3?) bands. The band which had length between 500~750 were very likely to be the successfully mutated (or the original template). The band between 250~500 might be the combination product of the two primer pairs. | + | From Double-primer PCR we got 2 (or 3?) bands. The band which had length between 500~750 were very likely to be the successfully mutated (or the original template). The band between 250~500 might be the combination product of the two primer pairs.<br> |
Next I would do gel extraction for the 500~750 band and do BbsI enzyme digestion to verify if the BbsI site was successfully mutated or not. | Next I would do gel extraction for the 500~750 band and do BbsI enzyme digestion to verify if the BbsI site was successfully mutated or not. | ||
| - | Gel extraction (13:53~15:20) | + | ==Gel extraction== |
| - | Tiangen Miniprep gel extraction kit (50 rxn) | + | (13:53~15:20 Aug 15th) |
| + | Tiangen Miniprep gel extraction kit (50 rxn)<br> | ||
As the Tiangen’s protocol instructed. | As the Tiangen’s protocol instructed. | ||
| - | Nanodrop test | + | ==Nanodrop test== |
| - | Band 700bp 400bp | + | {| class="table" |
| - | Conc (ng/ul) 54.7 73.2 | + | ! Band |
| - | 260/280 1.094 1.464 | + | ! 700bp |
| - | 260/230 0.634 0.445 | + | ! 400bp |
| + | |- | ||
| + | | Conc (ng/ul) | ||
| + | | 54.7 | ||
| + | | 73.2 | ||
| + | |- | ||
| + | | 260/280 | ||
| + | | 1.094 | ||
| + | | 1.464 | ||
| + | |- | ||
| + | | 260/230 | ||
| + | | 0.634 | ||
| + | | 0.445 | ||
| + | |} | ||
| - | BbsI enzyme | + | ==BbsI enzyme digestion== |
| - | Component Volume | + | (15:50~20:00, 4h10min) |
| - | NEB BbsI enzyme 0.2ul | + | {| class="table" |
| - | DNA (700bp band) 3ul | + | ! Component |
| - | 10X NEB buffer 2.1 1ul | + | ! Volume |
| - | ddH20 5.8ul | + | |- |
| - | Total 10ul | + | | NEB BbsI enzyme |
| + | | 0.2ul | ||
| + | |- | ||
| + | | DNA (700bp band) | ||
| + | | 3ul | ||
| + | |- | ||
| + | | 10X NEB buffer 2.1 | ||
| + | | 1ul | ||
| + | |- | ||
| + | | ddH20 | ||
| + | | 5.8ul | ||
| + | |- | ||
| + | | Total | ||
| + | | 10ul | ||
| + | |} | ||
75℃ 20min inactivation in dry bath | 75℃ 20min inactivation in dry bath | ||
| - | Gel electrophoresis (23:00~23:40): | + | ==Gel electrophoresis== |
| - | + | (23:00~23:40): | |
| - | Analysis: | + | {{SUSTC-Image|wiki/images/0/01/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-Double-Primer-PCR-8.17.jpg}} |
| - | After enzyme digestion, the band wasn’t cut into two bands (one about 440, another about 370). | + | '''Analysis:'''<br> |
| - | The next day I would do BbsI and BspEI double digestion to verify the results because BspEI would cut out a 99bp band. | + | After enzyme digestion, the band wasn’t cut into two bands (one about 440, another about 370). <br> |
| + | The next day I would do BbsI and BspEI double digestion to verify the results because BspEI would cut out a 99bp band. <br> | ||
P.S. The expected band length is 820bp, which seems to be inconsistent with the figure. However, comparing with the PCR result of mCherry, the band location seems to be the same, which means it was likely to be the problem of Marker. | P.S. The expected band length is 820bp, which seems to be inconsistent with the figure. However, comparing with the PCR result of mCherry, the band location seems to be the same, which means it was likely to be the problem of Marker. | ||
| - | Verification of the Point mutation | + | =Verification of the Point mutation= |
| - | + | ||
| - | + | ||
| - | + | ||
8.18 | 8.18 | ||
| - | Enzyme | + | ==Enzyme Digestion== |
| - | BbsI, BspEI double digestion (10:05~21:30) | + | ===BbsI, BspEI double digestion(10:05~21:30)=== |
| - | Component Volume | + | {| class="table" |
| - | BbsI 0.8ul | + | ! Component |
| - | BspEI 0.2ul | + | ! Volume |
| - | 700bp band 3ul | + | |- |
| - | 10X NEBuffer 3.1 1ul | + | | BbsI |
| - | ddH2O 5ul | + | | 0.8ul |
| - | Total 10ul | + | |- |
| - | + | | BspEI | |
| - | BspEI digestion (15:41~21:30) | + | | 0.2ul |
| - | Component Volume | + | |- |
| - | BbsI 0.2ul | + | | 700bp band |
| - | 700bp band 3ul | + | | 3ul |
| - | 10X NEBuffer 3.1 1ul | + | |- |
| - | ddH2O 5.8ul | + | | 10X NEBuffer 3.1 |
| - | Total 10ul | + | | 1ul |
| + | |- | ||
| + | | ddH2O | ||
| + | | 5ul | ||
| + | |- | ||
| + | | Total | ||
| + | | 10ul | ||
| + | |} | ||
| + | <span style="color: red">The activity of BbsI in NEBuffer 3.1 is only about 25%, so I increase the volumn the BbsI to 4X.</span> | ||
| + | ===BspEI digestion (15:41~21:30)=== | ||
| + | {| class="table" | ||
| + | ! Component | ||
| + | ! Volume | ||
| + | |- | ||
| + | | BbsI | ||
| + | | 0.2ul | ||
| + | |- | ||
| + | | 700bp band | ||
| + | | 3ul | ||
| + | |- | ||
| + | | 10X NEBuffer 3.1 | ||
| + | | 1ul | ||
| + | |- | ||
| + | | ddH2O | ||
| + | | 5.8ul | ||
| + | |- | ||
| + | | Total | ||
| + | | 10ul | ||
| + | |} | ||
| + | 80℃ inactivation, then immediately put into ice to avoid self-ligation.(21:40~22:40)<br> | ||
| + | ==Gel electrophoresis== | ||
| + | (22:50~23:20) | ||
| + | {{SUSTC-Image|wiki/images/e/ee/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-Double-Primer-PCR-Double-digestion-8.18.jpg}} | ||
| + | '''Analysis:'''<br> | ||
| + | The DNA might decompose due to too long inactivation time (1h at 80℃).<br> | ||
| + | However, Le Lin and Zeng Lidan thought high temp wouldn’t make DNA decompose. It was due to putting in ice immediately that makes DNA renaturation difficult.<br> | ||
| + | So, I decided to do enzyme digestion again without heat inactivation and do gel electrophoresis to see the results. I also will put the enzyme digestion system into 4℃ refrigerator overnight and do gel electrophoresis again the next day to verify if Le and Zeng’s idea was correct.<br> | ||
| - | + | ==Gel electrophoresis II== | |
| - | + | 8.19 | |
| - | + | {{SUSTC-Image|wiki/images/6/62/SUSTC-Shenzhen-mCherry-BbsI-pointmutation-Double-Primer-PCR-Double-digestion-8.19.jpg}} | |
| - | Analysis: | + | '''Analysis:'''<br> |
| - | + | From the gel electrophoresis result, Lin and Zeng’s idea was not correct because again no band is observed. BspEI digestion cut off a 99bp short segment and we can see the band moved quicker than BbsI band. The 273bp band was not observed in BspEI & BbsI double digestion, which means no BbsI site is in the PCR product. | |
| - | + | ||
| - | + | ||
| - | + | <span style="color: red">So the pointmutation was finally successful!!!</span> | |
| - | + | ||
| - | + | ||
| - | + | ||
| + | =References= | ||
| + | #[http://www.genomics.agilent.com/primerDesignProgram.jsp| QuikChange Primer Design] | ||
| + | #[http://www.genomics.agilent.com/article.jsp?pageId=384| QuikChange II Site-Directed Mutagenesis Kits - Details & Specifications] | ||
| + | # Mr. Wan Pei's Advice | ||
{{SUSTC-Shenzhen/main-content-end}} | {{SUSTC-Shenzhen/main-content-end}} | ||
{{SUSTC-Shenzhen/wiki-footer}} | {{SUSTC-Shenzhen/wiki-footer}} | ||
{{SUSTC-Shenzhen/themeJs}} | {{SUSTC-Shenzhen/themeJs}} | ||
Latest revision as of 14:11, 16 October 2014
mCherry BbsI point mutation
one step for constructing a universal usable gRNA-insertion plasmid
Contents |
By Yicong Tao
Plasmid used: pBX-084 PB5-HS4-TRE-mCherrynuc-2A-BlaloxN-GpA-HS4-PB3 (from Wei Huang’s lab). Total length is 6843bp.
Point mutation primer design:
Forward(5’-3’): cagcccatggttttcttctgcattacggggccg 33nt
Reverse(5’-3’): cggccccgtaatgcagaagaaaaccatgggctg 33nt
The primer pair is intended to eliminate the BbsI site in the mCherry because we need to use BbsI to insert our gRNA into our plasmid. Designed by Agilent Technologies point-mutation primer online design tools. The primers are complement with each other.
Agilent's Pointmutation method
8.13~8.14
PCR reaction
(12:06~14:XX 13th Aug, about 2h)
300ul, 50ul/reaction:
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 150ul | 1X |
| DNA template (251.4ng/ul) | 1.5ul | 1.257ng/ul |
| Primer F (50uM) | 3ul | 500nM |
| Primer R (50uM) | 3ul | 500nM |
| ddH20 | 142.5ul | - |
| Total | 300ul | - |
PCR reaction conditions:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 60 |
| 18 cycles | 98 | 10 |
| 58, 60, 61, 62, 63, 65 | 15 | |
| 72 | 360 | |
| Final extension | 72 | 120 |
| Hold | 4 | ∞ |
DpnI digestion
(13 Aug 15:05~19:06)
Add 1ul DpnI directly to the amplification system
Digest 4h
No termination
Stored at -20℃
Nanodrop test after digestion
| Anneal Temp (℃) | 58 | 60 | 61 | 62 |
|---|---|---|---|---|
| Conc (ng/ul) | 467.4 | 465.9 | 470.4 | 457.9 |
| 260/280 | 1.8 | 1.8 | 1.8 | 1.81 |
| 260/230 | 0.73 | 0.73 | 0.74 | 0.73 |
Bacteria transformation
(2014.8.13 22:08~2014.8.14 14:50)
Procedures:
- Immediately, place the tubes on ice, allow cell to thaw on ice, 10 min
- Check the cell to see if they have thawed, gently flick the cells 1-2 times to evenly resuspend the cells.
- Divide the competent cells into 6 tubes,50μL each tube, Mark them with 58, 60, 61, 62, 63, 65. Keep the bacteria in ice during the procedure.
- Add 5μL plasmid to each tube, shaking slightly
- Incubate on ice for 30min
- Heat shock, 42°C, 90s.
- Keep on ice, 2 min
- Add 200μl SOC medium to each tube, incubate 37°C, 200rpm, 45min
- Centrifuge, 4000rmp, 5min, RT. Discard most of the medium and reserve 50μL. Resuspend.
- Add 50μL bacteria to amp LB agar plate, distribute.
- Incubate the plates at 37°C, 16 hours.
Results:
For all 6 groups, no colony forms.
Gel electrophoresis
Making the agarose gel (1%)
| Component | Volume |
|---|---|
| TAE | 25ml |
| Agarose | 0.25g |
| Gene Green | 0.5ul |
Adding samples
| Component | Volume |
|---|---|
| PCR product | 1ul |
| 6X loading dye | 2ul |
| TAE | 9ul |
| Total | 12ul |
DNA marker (Takara DL5000, diluted): 10ul
Results:
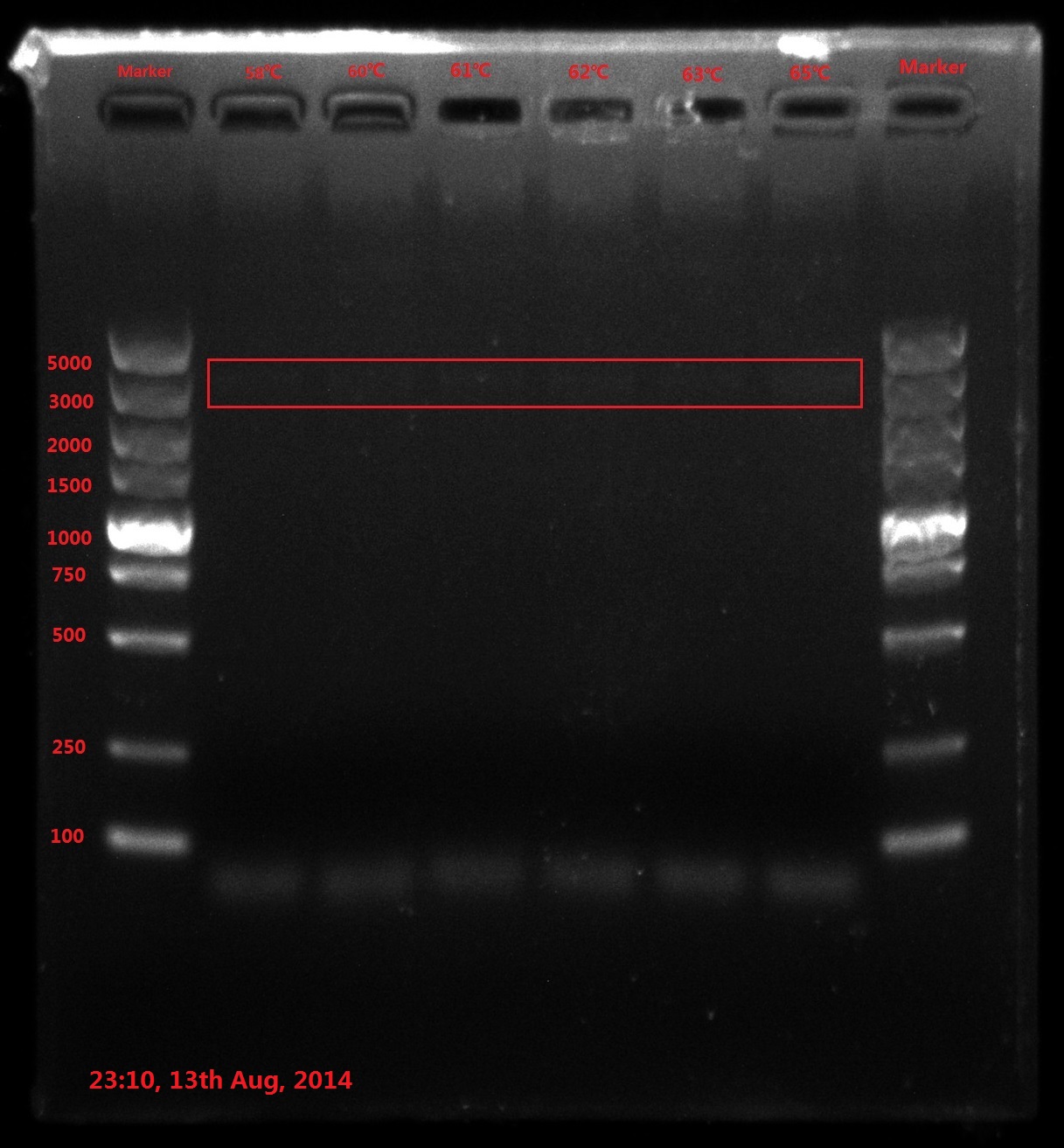
The result of electrophoresis is not clear, so I redid the gel electrophoresis with the same sample.
No wanted band (6843bp) is observed.
Discussion:
- PCR failed, so improving PCR conditions is the first choice. Try extending PCR cycles, denaturation time, annealing time and final extension time.
- Do reaction termination after DpnI enzyme digestion (80℃ 20min) next time because reactive enzyme may binding to the DNA substrate and interfere the transformation efficiency.
- The concentration of template DNA is also very essential, so using 2-step concentration ladder next time.
Agilent's Pointmutation method II
8.15
PCR
(23:35 14th Aug-08:30 15th Aug), about 2h. Next morning, check PCR machine was not at 4℃ hold conditions, and the heating stand was not cold
200ul, 50ul/reaction:
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 100ul | 1X |
| DNA template (251.4ng/ul) | 1.0ul (A,B), 2.0ul (C,D) | 1.257ng/ul (A,B), 2.514ng/ul (C,D) |
| Primer F (50uM) | 2ul | 500nM |
| Primer R (50uM) | 2ul | 500nM |
| ddH20 | 95ul | - |
| Total | 200ul | - |
PCR reaction conditions:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 60 |
| 25 cycles | 98 | 20 |
| 63 | 30 | |
| 72 | 420 | |
| Final extension | 72 | 600 |
| Hold | 4 | ∞ |
Nanodrop test
| Anneal Temp (℃) | A | B | C | D |
|---|---|---|---|---|
| Conc (ng/ul) | 474.2 | 492.0 | 466.7 | 445.9 |
| 260/280 | 1.82 | 1.81 | 1.81 | 1.81 |
| 260/230 | 0.72 | 0.73 | 0.72 | 0.70 |
Gel electrophoresis
Making the agarose gel (1%)
| Component | Volume |
|---|---|
| TAE | 25ml |
| Agarose | 0.25g |
| Gene Green | 0.5ul |
Adding samples
| Component | Volume |
|---|---|
| PCR product | 5ul |
| 6X loading dye | 2ul |
| TAE | 5ul |
| Total | 12ul |
DNA marker (Takara DL5000, diluted): 10ul
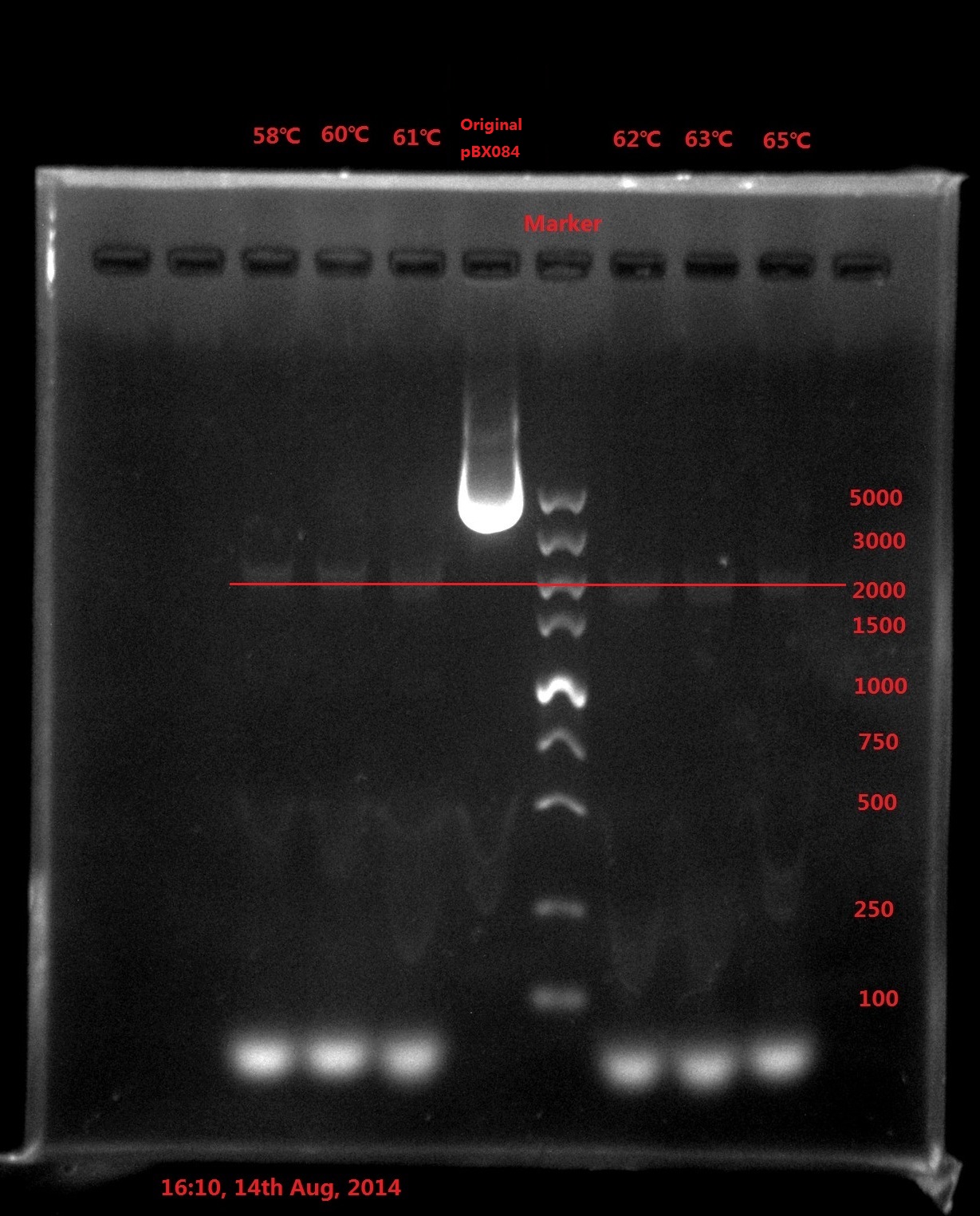
Results:
Analysis:
The band was in 2000bp location, which is far away from the original plasmid length (6800bp), which meant PCR didn’t got complete plasmid.
The possible reason might be:
- The primer forms dimers which strongly influence the efficiency of PCR (The smallest band is very light).
- DNA is a double helix, which means the plasmid double strand can also anneal with each other. The elongation time was very long, which left enough time for the plasmid double strand to anneal with each other and prohibit the PCR process.
Solution:
- Redesign the primer so that the primer pairs are not totally complementary with each other. Put the point mutation point near the 5’ end.
- Use other PCR methods.
So I decided to change the PCR method.
2-pair primers PCR
I had two pairs of primers: one is for mCherry (called A), another is for point mutation (called B). I then used the forward primer of A to pair with the reverse primer of B, so did the forward primer of B with the reverse primer of A. Thus, I would get 2 short segments which had a small complementary region. Then, I added the two segments together and let them do PCR using each other as the template and primer in order to get the final whole length PCR product.
mCherry primer design:
Forward(5’-3’): TCGTTTAGTGAACCGTCAG 19nt
Reverse(5’-3’): cgaggaattcctattaTTCCTCTGCCCTCACTAG 34nt
PCR
50ul, 25ul/reaction:
Reaction 1:
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 25ul | 1X |
| pBX084 diluted (25.14ng/ul) | 2.0ul | 1.257ng/ul |
| mCherry Primer F (50uM) | 0.5ul | 500nM |
| Pointmutation Primer R (50uM) | 0.5ul | 500nM |
| ddH20 | 22ul | - |
| Total | 50ul | - |
Reaction 2:
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 25ul | 1X |
| pBX084 diluted (25.14ng/ul) | 2.0ul | 1.257ng/ul |
| mCherry Primer R (50uM) | 0.5ul | 500nM |
| Pointmutation Primer F (50uM) | 0.5ul | 500nM |
| ddH20 | 22ul | - |
| Total | 50ul | - |
PCR reaction conditions:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 60 |
| 25 cycles | 98 | 20 |
| 61 | 15 | |
| 72 | 20 | |
| Final extension | 72 | 300 |
| Hold | 4 | ∞ |
Gel electrophoresis
Making the agarose gel (1%)
| Component | Volume |
|---|---|
| TAE | 25ml |
| Agarose | 0.25g |
| Gene Green | 0.5ul |
Adding samples
| Component | Volume |
|---|---|
| PCR product | 10ul |
| 6X loading dye | 2ul |
| Total | 12ul |
DNA marker (Takara DL5000, diluted): 30ul

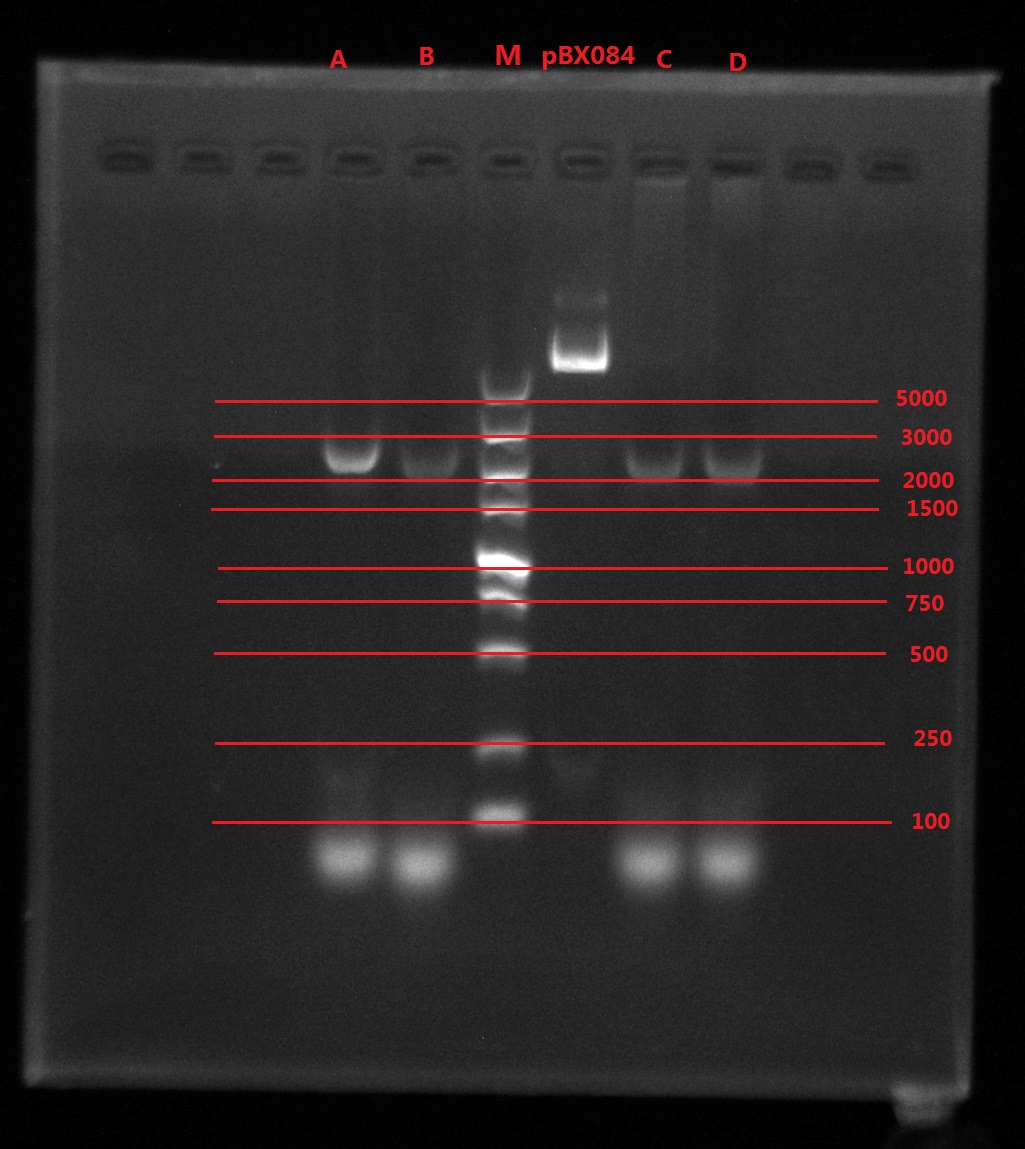
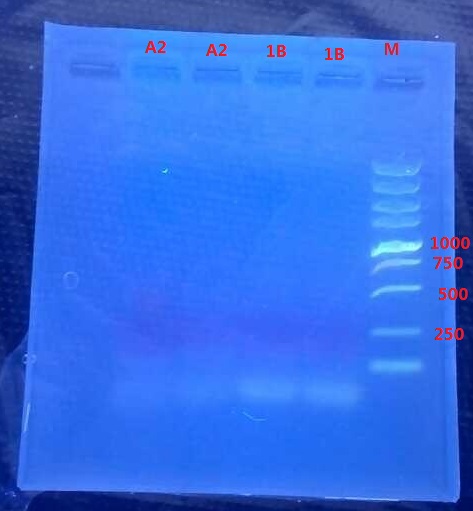
Results:
A2: reaction 1 (A: mCherry primer F; 2: Pointmutation Primer R)
1B: reaction 2 (B: mCherry primer R; 1: Pointmutation Primer F)
Analysis:
The predicting results is 448bp band in A2 and 372bp band in 1B (almost equally separate the mCherry). But the band in A2 was in 800bp and no band is observed in 1B. The possible reasons might be:
- I added wrong primer pairs: two mCherry primers for A2 and two pointmutation primers for 1B. So that in A2 I got mCherry and in 1B I got primer dimer.
- The elongation time might be too long.
So I redid the 2-pari primers PCR with modified PCR reaction conditions.
PCR II
Reaction conditions:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 60 |
| 25 cycles | 98 | 20 |
| 61 | 15 | |
| 72 | 15 | |
| Final extension | 72 | 120 |
| Hold | 4 | ∞ |
Gel electrophoresis:
 Results:
Results:
No band is observed.
Analysis:
- The elongation time might be too short…
- The annealing time for 1B might be wrong.
2-pari primers PCR II
8.16 10:25am~11.25am
I set a temperature gradient (45, 48, 51, 54, 57, 60℃) without shortening the elongation time.
PCR
150ul, 25ul/reaction:
Reaction 1:
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 25ul | 1X |
| pBX084 diluted (25.14ng/ul) | 0.5ul | 12.57ng |
| Pointmutation Primer F (50uM) | 1ul | 1000nM |
| ddH20 | 23ul | - |
| Total | 49.5ul | - |
Reaction 2:
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 25ul | 1X |
| pBX084 diluted (25.14ng/ul) | 0.5ul | 12.57ng |
| Pointmutation Primer R (50uM) | 1ul | 1000nM |
| ddH20 | 23ul | - |
| Total | 49.5ul | - |
PCR reaction conditions:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 60 |
| 25 cycles | 98 | 20 |
| 63 | 15 | |
| 72 | 5min | |
| Final extension | 72 | 10min |
| Hold | 4 | ∞ |
Gel electrophoresis
11:50am ~12:25pm
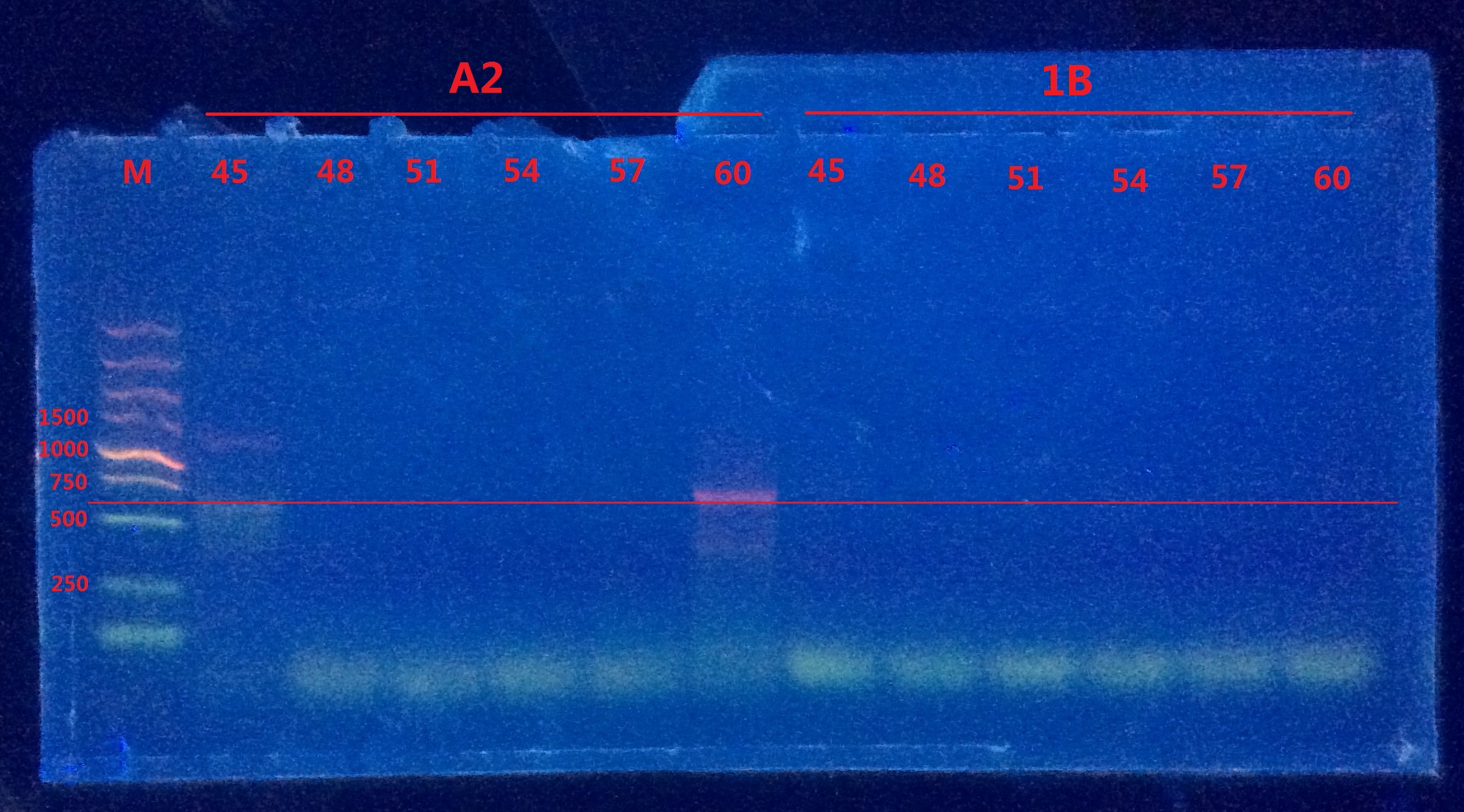 Unfortunately, only in A2 did I observe the band. I doubt the annealing temperature was not high enough because strong primer dimers forms in 1B group. So I redid the 2-pair primers PCR again with even higher temperature gradient (62,63,64,65,66,67℃).
Unfortunately, only in A2 did I observe the band. I doubt the annealing temperature was not high enough because strong primer dimers forms in 1B group. So I redid the 2-pair primers PCR again with even higher temperature gradient (62,63,64,65,66,67℃).
Gel electrophoresis II
 Results:
Results:
Only vague bands were observed in A2. Still no distinct bands observed in 1B.
Analysis:
- The reverse primer of pointmutation might be very easy to form dimer with the forward primer of mCherry, which led to the failure of PCR.
- The compatibility of the two separate primers might be very bad.
I decided to try another methods.
Half-plasmid PCR & Double primer PCR
Half-plasmid PCR
First, only add one direction primer so I could get half linear plasmid. After that, I mix the two DNA together to get the complete DNA.
50ul, 25ul/reaction:
Reaction 1
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 25ul | 1X |
| pBX084 diluted (25.14ng/ul) | 0.5ul | 12.57ng |
| Pointmutation Primer F (50uM) | 1ul | 1000nM |
| ddH20 | 23ul | - |
| Total | 49.5ul | - |
Reaction 2
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 25ul | 1X |
| pBX084 diluted (25.14ng/ul) | 0.5ul | 12.57ng |
| Pointmutation Primer R (50uM) | 1ul | 1000nM |
| ddH20 | 23ul | - |
| Total | 49.5ul | - |
PCR reaction conditions:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 60 |
| 25 cycles | 98 | 20 |
| 63 | 15 | |
| 72 | 5min | |
| Final extension | 72 | 10min |
| Hold | 4 | ∞ |
Double primer PCR
Mix the two pairs of primer together but the density of the pointmutation primer is only 1/10 of the mCherry primer. So the pointmutation primer will be consumed very quickly in the first several rounds.
150ul, 25ul/reaction:
| Component | Volume | Final Concentration |
|---|---|---|
| Q5 master mix 2X | 75ul | 1X |
| pBX084 diluted (25.14ng/ul) | 1ul | 25.14ng |
| mCherry Primer F&R (50uM) | 2ul | 6666.7nM |
| Pointmutation Primer R (50uM) | 0.2ul | 666.7nM |
| ddH20 | 69.6ul | - |
| Total | 150ul | - |
PCR reaction conditions:
| Step | Temp (℃) | Time (s) |
|---|---|---|
| Initial denaturation | 98 | 60 |
| 25 cycles | 98 | 20 |
| 45,48,52,55,58,61 | 20 | |
| 72 | 20 | |
| Final extension | 72 | 5min |
| Hold | 4 | ∞ |
The two PCR methods proceed simultaneously from 17:00 to 19:30, then reserved in 4℃.
Gel electrophoresis
8.17
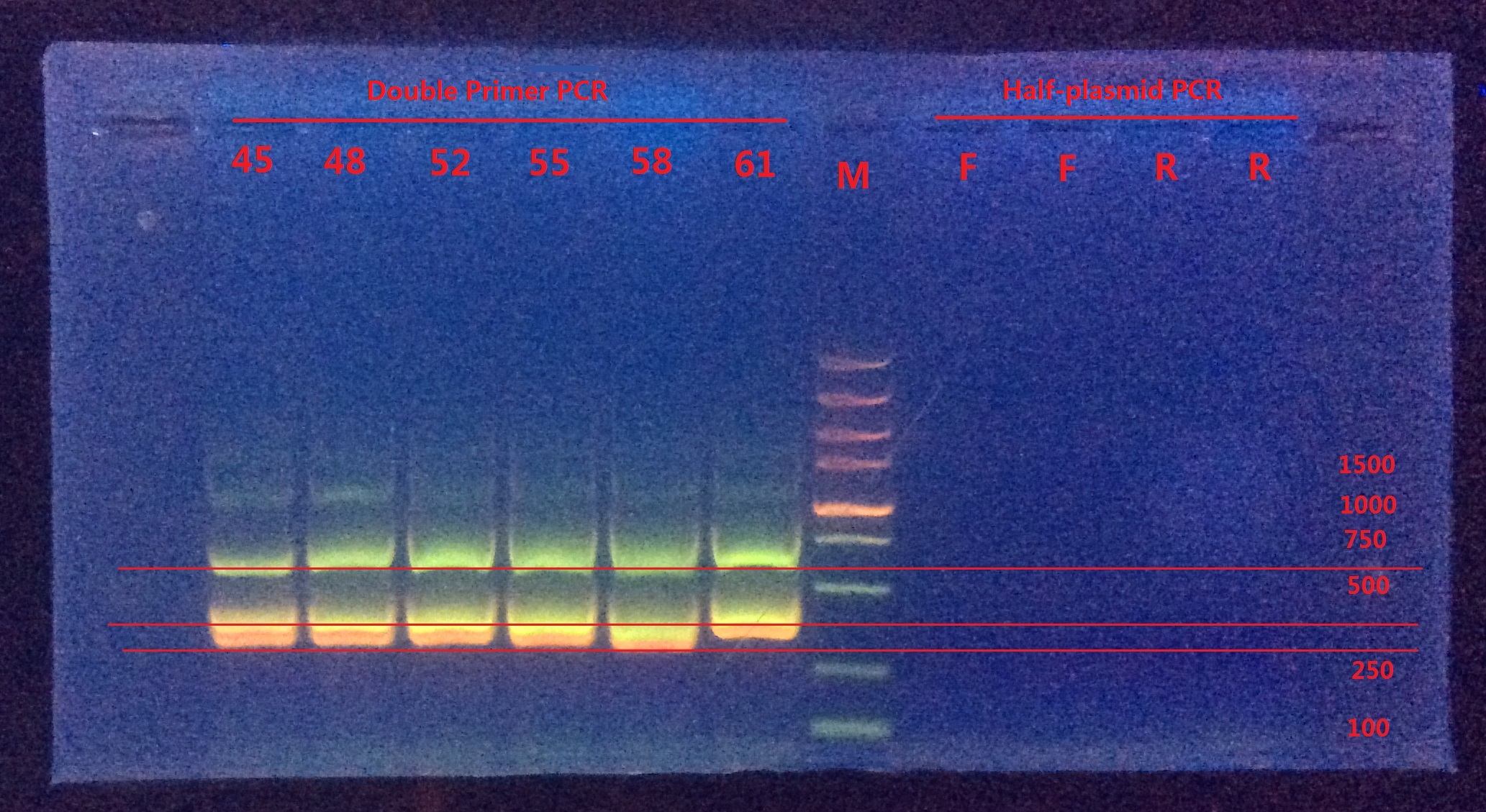 Analysis:
Analysis:
From Double-primer PCR we got 2 (or 3?) bands. The band which had length between 500~750 were very likely to be the successfully mutated (or the original template). The band between 250~500 might be the combination product of the two primer pairs.
Next I would do gel extraction for the 500~750 band and do BbsI enzyme digestion to verify if the BbsI site was successfully mutated or not.
Gel extraction
(13:53~15:20 Aug 15th)
Tiangen Miniprep gel extraction kit (50 rxn)
As the Tiangen’s protocol instructed.
Nanodrop test
| Band | 700bp | 400bp |
|---|---|---|
| Conc (ng/ul) | 54.7 | 73.2 |
| 260/280 | 1.094 | 1.464 |
| 260/230 | 0.634 | 0.445 |
BbsI enzyme digestion
(15:50~20:00, 4h10min)
| Component | Volume |
|---|---|
| NEB BbsI enzyme | 0.2ul |
| DNA (700bp band) | 3ul |
| 10X NEB buffer 2.1 | 1ul |
| ddH20 | 5.8ul |
| Total | 10ul |
75℃ 20min inactivation in dry bath
Gel electrophoresis
(23:00~23:40):
 Analysis:
Analysis:
After enzyme digestion, the band wasn’t cut into two bands (one about 440, another about 370).
The next day I would do BbsI and BspEI double digestion to verify the results because BspEI would cut out a 99bp band.
P.S. The expected band length is 820bp, which seems to be inconsistent with the figure. However, comparing with the PCR result of mCherry, the band location seems to be the same, which means it was likely to be the problem of Marker.
Verification of the Point mutation
8.18
Enzyme Digestion
BbsI, BspEI double digestion(10:05~21:30)
| Component | Volume |
|---|---|
| BbsI | 0.8ul |
| BspEI | 0.2ul |
| 700bp band | 3ul |
| 10X NEBuffer 3.1 | 1ul |
| ddH2O | 5ul |
| Total | 10ul |
The activity of BbsI in NEBuffer 3.1 is only about 25%, so I increase the volumn the BbsI to 4X.
BspEI digestion (15:41~21:30)
| Component | Volume |
|---|---|
| BbsI | 0.2ul |
| 700bp band | 3ul |
| 10X NEBuffer 3.1 | 1ul |
| ddH2O | 5.8ul |
| Total | 10ul |
80℃ inactivation, then immediately put into ice to avoid self-ligation.(21:40~22:40)
Gel electrophoresis
(22:50~23:20)
 Analysis:
Analysis:
The DNA might decompose due to too long inactivation time (1h at 80℃).
However, Le Lin and Zeng Lidan thought high temp wouldn’t make DNA decompose. It was due to putting in ice immediately that makes DNA renaturation difficult.
So, I decided to do enzyme digestion again without heat inactivation and do gel electrophoresis to see the results. I also will put the enzyme digestion system into 4℃ refrigerator overnight and do gel electrophoresis again the next day to verify if Le and Zeng’s idea was correct.
Gel electrophoresis II
8.19
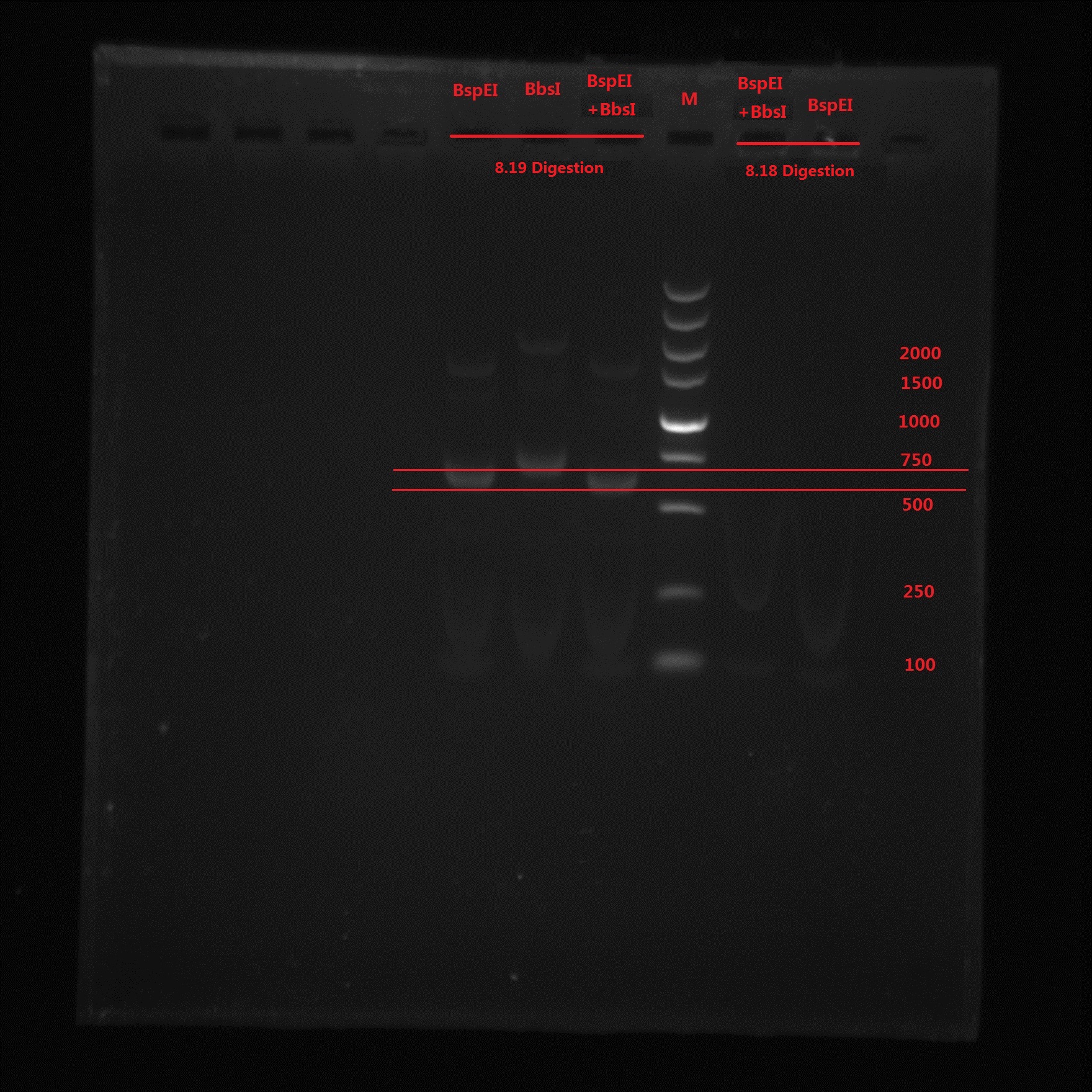 Analysis:
Analysis:
From the gel electrophoresis result, Lin and Zeng’s idea was not correct because again no band is observed. BspEI digestion cut off a 99bp short segment and we can see the band moved quicker than BbsI band. The 273bp band was not observed in BspEI & BbsI double digestion, which means no BbsI site is in the PCR product.
So the pointmutation was finally successful!!!
References
- [http://www.genomics.agilent.com/primerDesignProgram.jsp| QuikChange Primer Design]
- [http://www.genomics.agilent.com/article.jsp?pageId=384| QuikChange II Site-Directed Mutagenesis Kits - Details & Specifications]
- Mr. Wan Pei's Advice