|
|
| (74 intermediate revisions not shown) |
| Line 3: |
Line 3: |
| | <title>Cooper Union 2014 iGEM</title> | | <title>Cooper Union 2014 iGEM</title> |
| | <body> | | <body> |
| - |
| |
| | <div class="parent-container"> | | <div class="parent-container"> |
| | | | |
| Line 10: |
Line 9: |
| | <ul class="menu"> | | <ul class="menu"> |
| | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union"> Home </a> </li> | | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union"> Home </a> </li> |
| - | <li class="menu"> Project | + | <li class="menu-project"> Project |
| | + | <ul class="menu"> |
| | + | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/TdT_project">De Novo Synthesis </a></li> |
| | + | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Telomere_project">Programmable Lifespan Timer</a> </li> |
| | + | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Biohack_project">Biohacker Kit </a></li> |
| | + | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Hardware">OpenSource Hardware </a> </li> |
| | + | <!--<li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Modeling">Modeling </a> </li>--> |
| | + | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Parts">BioBrick Parts </a></li> |
| | + | </ul> |
| | + | </li> |
| | + | <li class="menu-safety"> Social |
| | <ul class="menu"> | | <ul class="menu"> |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/TdT_project">De Novo Synthesis </a></li> | + | <li class="menu"><a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Safety">Safety</a> </li> |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Telomere_project">Programmable Lifespan</a> </li> | + | <li class="menu"><a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Ethics">Ethics</a> </li> |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Biohack_project">Biohacker Kit </a></li> | + | <li class="menu"><a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Outreach">Outreach </a></li> |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Hardware">OpenSource Hardware </a> </li>
| + | |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Parts">BioBrick Parts </a></li>
| + | |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Modeling">Modeling </a> </li>
| + | |
| | </ul> | | </ul> |
| | </li> | | </li> |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Safety">Safety</a> </li> | + | <li class="menu-notebook"> Notebook |
| - | <li class="menu"><a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Outreach"> Outreach </a></li>
| + | |
| - | <li class="menu"> Notebook
| + | |
| | <ul class="menu"> | | <ul class="menu"> |
| | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Protocols">Commonly Used Protocols </li> | | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Protocols">Commonly Used Protocols </li> |
| Line 28: |
Line 32: |
| | </ul> | | </ul> |
| | </li> | | </li> |
| - | <li class="menu"> Team | + | <li class="menu-team"> Team |
| | <ul class="menu"> | | <ul class="menu"> |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/biofanfic">Members </a> </li> | + | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/biofanfic">Members </a> </li> |
| - | <li class="menu"> <a class="menu" href="https://2014.igem.org/wiki/index.php?title=Team:Cooper_Union/Attributions"> Attributions </a> </li> | + | <li class="menu"> <a class="menu" href="https://2014.igem.org/Team:Cooper_Union/Attributions"> Attributions </a> </li> |
| | <li class="menu"> <a class="menu" href="https://igem.org/Team.cgi?id=1354"> Official Team Profile </a> </li> | | <li class="menu"> <a class="menu" href="https://igem.org/Team.cgi?id=1354"> Official Team Profile </a> </li> |
| | </ul> | | </ul> |
| Line 37: |
Line 41: |
| | </ul> | | </ul> |
| | <a class="menu" href="http://www.igem.org" border="0"> | | <a class="menu" href="http://www.igem.org" border="0"> |
| - | <img src="https://static.igem.org/mediawiki/igem.org/f/f2/Igemfooterlogo.png" class="menu-top-logo" /> | + | <img src="https://static.igem.org/mediawiki/igem.org/9/94/Igemlogo_officiallogo.png" class="menu-top-logo" /> |
| | </a> | | </a> |
| | | | |
| | </div> | | </div> |
| - | | + | <br> |
| | + | <hr> |
| | + | <br> |
| | + | <div class="text"> |
| | <!-----Add content below----> | | <!-----Add content below----> |
| - | <b>De novo Enzyme Mediated Oligonucleotide Synthesizer</b>
| |
| - | <br><br>
| |
| | | | |
| | | | |
| - | De novo synthesis is a way of creating DNA oligonucleotides without the need of a template strand. Since the conventional method is expensive, time consuming, and inefficient, our team wants to focus on the ways to minimize the time and money required for DNA synthesis and to allow the labs to produce oligonucleotides easily without ordering. Here we introduce the De novo Enzyme Mediated Oligonucleotide Synthesizer (DEMOS); a programmable enzyme based, template-free, synthesizer for nucleic acid polymers. | + | |
| - | Our eventual goal will be to build a microfluidic de novo synthesizer that will allow laboratories, from academia to DIYBio community labs to rapidly and economically synthesize any strand of DNA for their projects.We hope that this system will become the key platform that bridges the <em>in silico</em> to <em>in vitro</em> gap in the design-test-build cycle of DNA synthesis and experimentation. | + | <br><br> |
| | + | <div class="center"><h1>De novo Enzyme Mediated Oligonucleotide Synthesizer</h1></div> |
| | + | <br> |
| | + | |
| | + | |
| | + | De novo synthesis is a way of creating DNA oligonucleotides without the need of a template strand. Since the conventional method is expensive, time consuming, and inefficient, our team has focused on minimizing the time and money required for DNA synthesis while allowing labs to produce oligonucleotides easily without ordering. Here we introduce the De novo Enzyme Mediated Oligonucleotide Synthesizer (DEMOS); a programmable enzyme based, template-free, synthesizer for nucleic acid polymers. Our eventual goal will be to build a microfluidic De novo synthesizer that will allow laboratories, from academia to DIYBio community labs to rapidly and economically synthesize any strand of DNA for their projects. We hope that this system will become the key platform that bridges the <em>in silico</em> to <em>in vitro</em> gap in the design-test-build cycle of DNA synthesis and experimentation. |
| | <BR><BR> | | <BR><BR> |
| | | | |
| - | Our system is fundamentally based on two key technologies, one practical and the other theoretical: 1) the development of deoxynucleotide triphosphate (dNTP) substrates with 3' reversible protective groups for "sequencing by synthesis" and "hot start" PCR technologies and the published theoretical model of directed template-free synthesis of DNA using the enzyme terminal deoxynucleotidyl transferase. This enzyme that has the ability to add nucleotides to the 3' ends of DNA, preferable 3' overhangs, in a template-free manner. (TdT). | + | Our system is fundamentally based on two key technologies, one practical and the other theoretical: |
| | + | <ol> |
| | + | <li>The development of deoxynucleotide triphosphate (dNTP) substrates with 3' reversible protective groups for "sequencing by synthesis" and "hot start" PCR technologies</li> |
| | + | <li>The published theoretical model of directed template-free synthesis of DNA using the enzyme terminal deoxynucleotidyl transferase (TdT). This enzyme that has the ability to add nucleotides to the 3' ends of DNA, preferable 3' overhangs, in a template-free manner. </li> |
| | + | </ol> |
| | + | |
| | <BR><BR> | | <BR><BR> |
| - | In the general scheme shown in Figure 1 below, a growing DNA chain possessing a free 3'OH end has one incoming 3'-RPdNTP (reverse protective group dNTP) enzymatically added by TdT at the permissive temperature of 37 degrees Celsius. This is followed by a wash step to remove unincorporated 3'RPdNTPs and PPi (pyrophosphate). Deprotection is achieved by raising the temperature to 95 degrees Celsius, in the case of heat labile protective groups, or pulsing with ultra-violet light when using photolabile protective groups. This is followed by another wash that removes decoupled protective groups, thereby resetting the system to begin another cycle of addition. | + | In the general scheme shown below in Figure 1, an incoming 3'-RPdNTP (reverse protective group dNTP) is added to a free 3'OH of a growing DNA chain by the enzyme TdT at 37° Celsius. This is followed by a wash step where unincorporated 3'RPdNTPs and PPi (pyrophosphate) are removed. For heat labile 3'-RPdNTP’s deprotection is achieved by raising the temperature to 95° Celsius, while photolabile 3'-RPdNTP’s are deprotected by pulses of ultra-violet light. This is followed by another wash step where the decoupled protective groups are removed, thereby resetting the system to begin another cycle of addition. |
| | <BR><BR> | | <BR><BR> |
| - | <img src= "https://static.igem.org/mediawiki/2014/1/15/CooperUnionTdT1.jpg"/>
| + | <div class="center"> |
| | + | <img src= "https://static.igem.org/mediawiki/2014/1/15/CooperUnionTdT1.jpg" width="500"/><br> |
| | + | <span>Figure 1. General DNA De novo Synthesis Scheme</span></div> |
| | + | <BR><BR><br> |
| | + | The deprotection reaction of the 3'-RPdNTP’s is shown in Figure 2a. The structures of two heat labile 3' protective group dNTPs that are commercially available from TriLink Biotechnologies (www.trilinkbiotech.com) are shown in Figure 2b . For our proof of concept experiments, 3'-TBE-dNTP’s were used, as it has a faster rate of deprotection at 95° Celsius; 3'-TBE-dNTP t 1/2 + 5 minutes, 3'-THF-dNTP t 1/2= 90 minutes) |
| | <BR><BR> | | <BR><BR> |
| - | Figure 2 below summarizes the reaction and structure of two heat labile 3'protective group dNTPs that are commercially available from TriLink Biotechnologies (www.trilinkbiotech.com). For our proof of concept experiments, 3'-TBE-dNTP was used, as it has a faster rate of deprotection at 95 degrees Celsius; 3'-TBE-dNTP t 1/2 + 5 minutes, 3'-THF-dNTP t 1/2= 90 minutes)
| + | <div class="center"> |
| | + | <img src= "https://static.igem.org/mediawiki/2014/1/13/CooperUnionTdT4.jpg" height="150" /><br> |
| | + | <span>Figure 2 a. General Deprotection of Protected Nucleotide b. Thermolabile Protective Groups</span></div> |
| | <BR><BR> | | <BR><BR> |
| - | <img src= "https://static.igem.org/mediawiki/2014/4/4d/CooperUnionTdT2.jpg" /> | + | <br><br> |
| | + | |
| | + | <div class="center"> |
| | + | <h2>Advantages of Our System vs. Phosphoramidite Mediated Oligonucleotide Synthesis</h2><br> |
| | + | <img src= "https://static.igem.org/mediawiki/2014/8/8a/CooperUnionTdT3.png" width="650" /><br> |
| | + | <span>Figure 3. General Phosphoramidite Synthesis Cycle Scheme</span></div> |
| | <BR><BR> | | <BR><BR> |
| - | <b> Advantages of De novo Enzyme Mediated DNA Synthesis </b> | + | <br><br> |
| | | | |
| | + | Overall, there are several notable advantages of our system over the Phosphoramidite system. Our system is substantially more eco-friendly, as some of the reagents used in the Phosphoramidite system are considered toxic. For instance, Palladium (Pd) complex is used to remove the protecting group from the dNTPs. Pd is considered to have low toxicity because the absorbency rate in the body is relatively low. However when the Pd complex is used as a reagent, it is possible for its toxicity to increase. There have been cases where it has caused acute toxic effects in lab mice, including mutagenesis to their isolated hearts. Another toxic reagent used is Dichloroacetic acid (DCA). DCA has been listed by the Environmental Protection Agency as a cancer-causing agent in humans and is also known to cause infertility in men. Additionally, long term use is associated with increased risk of liver cancer. Thus the use of these reagents is not cost effective since many precautions must be taken for their use. Permits must be obtained for their use, proper lab and safety equipment must be installed, safety protocols must be followed, and the reagents must be disposed of properly. Another advantage our system is that it is faster overall. As shown above in Figure 3, one cycle for the Phosphoramadite system is comprised of four main steps, while ours has only two. This greatly increases the kinetics of the system since the time it takes to complete a full synthesis is equivalent to the time that it takes to complete one cycle multiplied by the number of base pairs that will be synthesized. <br><br> |
| | | | |
| - | <b>pH Regulated De novo Enzyme Mediated DNA Synthesis </b> | + | <div class="center"> |
| - | In the controllable system proposed by Ud-Dean, the reversibility of a 3' acetyl protective group was achieved by lowering pH during each cycle and thereby activating a deacetylase enzyme that is active at lower pH. During this step, TdT is also inactived by the shift to lower pH, thereby preventing the addition of extra nucleotides | + | <h2>Advantages of Our System vs. pH Regulated De novo Enzyme Mediated DNA Synthesis</h2> |
| | + | </div> |
| | + | <BR> |
| | + | In the controllable system proposed by Ud-Dean, the reversibility of a 3' acetyl protective group was achieved by lowering pH during each cycle and thereby activating a deacetylase enzyme that is active at lower pH. During this step, TdT is also inactivated by the shift to lower pH, thereby preventing the addition of extra nucleotides. |
| | + | <BR><BR> |
| | + | <div class="center"><img src= "https://static.igem.org/mediawiki/2014/7/7b/CooperUnionTdT5.png" width="550" /><br> |
| | + | <span>Figure 4. General pH Regulated De novo Enzyme Mediated DNA Synthesis Cycle Scheme</span></div> |
| | + | <BR><BR> |
| | + | Our system simplifies this novel approach by using thermolabile reversible 3' protective groups. Since our system uses heat to deprotect the dNTPs, only one step is needed instead of two, decreasing the time that is needed to add one base pair. This greatly increases the kinetics of the system since the time it takes to complete a full synthesis is equivalent to the time that it takes to complete one cycle multiplied by the number of base pairs that will be synthesized. |
| | + | <BR><BR> |
| | | | |
| - | Our system simplifies this novel approach by thermolabile reversible 3' protective groups, thereby combining the
| |
| | | | |
| | <BR><BR> | | <BR><BR> |
| - | <b>Phosphoramidite Mediated Oligonucleotide Synthesis </b> | + | <div class="center"><h2>Results</h2> |
| - |
| + | <br><br><img src="https://static.igem.org/mediawiki/2014/0/09/CU_1017_TdT.png" width="400" /><br> |
| - | | + | <span>Figure 5. TdT Enzyme Functional Assay</span></div><br><br> |
| | + | <br>As seen from the polyacrylamide gel shown above in Figure 5, the TdT enzyme expressed in the Rosetta cells using our genetically engineered plasmid works as intended; base pairs are appended to oligos by the enzyme. However, there is a difference between the activity of our enzyme compared to the commercially obtained TdT. The majority of the oligos have only a few base pairs added by our expressed TdT while the lane of the commercial TdT is a smear of oligos, suggesting the commercial TdT has a greater reaction rate. But it can also be attributed to the fact that the 0.5 U/μL concentration of our expressed TdT is an overestimation. The TdT concentration of the reaction with our expressed TdT is less than that of the commercial TdT. |
| | + | <br><br> |
| | + | There is also a large gap between the oligos with only a few bases added and those with 30+ bases added. This suggests that our expressed TdT has a higher processivity than that of commercially obtained TdT. This might be due to subtle sequence differences such as the His tag on our TdT sequence. |
| | | | |
| | + | <br><br> |
| | + | <div class="center"><h2>Potential Errors in the Synthesis System</h2></div> |
| | + | <BR> |
| | | | |
| | + | During each cycle there are independent sources of error that can affect the overall efficiency of the reaction. The most obvious errors include: |
| | + | <ol> |
| | + | <li>Incomplete enzymatic addition of 3' reversibly protected dNTPs by TdT during the nucleotide incorporation step.</li><li>Incomplete hydrolysis of 3' protective group during heating or other treatment.</li> |
| | + | <li>Incomplete removal of unincorporated 3' reversibly protected nucleotides after each incorporation step.</li> |
| | + | <li>Spontaneous hydrolysis of 3' protective groups prior to intended deprotection. Each error, if not sufficiently addressed would compound the likelihood of misincorporated nucleotides and a reduction of overall yield. </li> |
| | + | </ol> |
| | + | <BR><BR> |
| | + | |
| | + | For example, during error scenario 1, oligonucletide chains that have not had a 3' reversibly protective group nucleotide added during an incorporation step, will, during the subsequent step after deprotection, be potential substrates for the addition of the next incoming nucleotide. This would lead to the base deletion. During error scenario 2, the failure to remove a protective group would lead to a population of oligonucleotides that would fail to add a nucleotide during the next subsequent addition of incoming nucleotides, leading to a base deletion at this step. For error scenario 3, the presence of unremoved, unreacted nucletides would lead to misincorporation and hence base substitutions during each subsequent stage of oligonucleotide elongation. During Scenario 4, the presence of dNTPs without 3' protective groups would lead to the incorporation of multiple dNTPs, resulting in homopolymeric stretches of nucleotides in the growing oligonucleotide chain. This would lead to insertion mutations. For a truly reliable system to be built, these and potentially other sources of error will need to be adequately countered. |
| | + | <BR><BR> |
| | | | |
| | | | |
| - |
| |
| | <br><br> | | <br><br> |
| - | <b>Future Improvements to the De novo Oligonucleotide Synthesizer</b> | + | <div class="center"><h2>Future Improvements to the De novo Oligonucleotide Synthesizer</h2></div> |
| | <br><br> | | <br><br> |
| - | In our present configuration, full length recombinant bovine TdT was used in the reaction. This particular enzyme denatures in the presence of elevated temperatures, thereby necessitating the repeated addition of enzyme during each step of nucleotide addition after blocking group removal via 95 degrees C heat pulse. Therefore it would be useful to either isolate a naturally occurring variant of TdT or similar enzyme, that is heat tolerant, most likely from a thermophylic organism, or engineer such a variant. | + | In our present configuration, full length recombinant bovine TdT was used in the addition of thermolabile dNTPs to single stranded oligos. This particular enzyme denatures under elevated temperatures. Therefore, after the blocking group is removed from the dNTP at 95°C, it is necessary to add more of the enzyme. As a result, it would be useful to either isolate a naturally occurring variant of TdT or a similar enzyme that is resistant to denaturation under high temperature conditions. This type of enzyme might either be found in a thermophilic organism or synthetically engineered in a laboratory. Another potential improvement would be to construct truncated and other variants of engineered TdT that would incorporate modified dNTPs at a faster rate. Lastly, our current system could be improved by switching from commercially available thermolabile nucleotides to photolabile nucleotides. This should improve the system since temperature ramp up and down cycles are avoided, creating a faster reaction cycle and eliminating the need to re-add the TdT enzyme at the beginning of each cycle. We are presently exploring these improved features. |
| - | | + | |
| - | Another potential course of improvement would be to construct truncated and other variants of engineered TdT that exhibit faster kinetics of modified nucleotide incorporation. | + | |
| - | | + | |
| - | Lastly, our present system used commercially available nucleotides that had heat labile 3' protective groups. By switching to photolabile nucleotides, the system should perform better by exhibiting an overall faster reaction cycle, since temperature ramp up and down cycles are avoided and no extra TdT enzyme would need to be added at the beginning of each cycle, as heat denaturation would also be avoided. We are presently exploring these improved features. | + | |
| - |
| + | |
| | <br><br> | | <br><br> |
| | | | |
| | | | |
| | | | |
| - | <B>Potential Errors in the Synthesis System</B>
| |
| - | <BR><BR>
| |
| | | | |
| - | During each cycle there are independent sources of error that can affect the overall efficiency of the reaction. The most obvious errors include: 1) Incomplete enzymatic addition of 3' reversibly protected dNTPs by TdT during the nucleotide incorporation step. 2) Incomplete hydrolysis of 3' protective group during heating or other treatment. 3) Incomplete removal of unincorporated 3' reversibly protected nucleotides after each incorporation step. 4) Spontaneous hydrolysis of 3' protective groups prior to intended deprotection. Each error, if not sufficiently addressed would compound the likelihood of misincorporated nucleotides and a reduction of overall yield.
| |
| - | <BR><BR>
| |
| - |
| |
| - | For example, during error scenario 1, oligonucletide chains that have not had a 3' reversibly protective group nucleotide added during an incorporation step, will, during the subsequent step after deprotection, be potential substrates for the addition of the next incoming nucleotide. This would lead to the base deletion. During error scenario 2, the failure to remove a protective group would lead to a population of oligonucleotides that would fail to add a nucleotide during the next subsequent addition of incoming nucleotides, leading to a base deletion at this step. For error scenario 3, the presence of unremoved, unreacted nucletides would lead to misincorporation and hence base substitutions during each subsequent stage of oligonucleotide elongation. During Scenario 4, the presence of dNTPs without 3' protective groups would lead to the incorporation of multiple dNTPs, resulting in homopolymeric stretches of nucleotides in the growing oligonucleotide chain. This would lead to insertion mutations. For a truly reliable system to be built, these and potentially other sources of error will need to be adequately countered.
| |
| | <BR><BR> | | <BR><BR> |
| | | | |
| | <b>References</b> | | <b>References</b> |
| - | <BR><BR> | + | <ol> |
| - | 1.Minhaz Ud-Dean, S.M. (2008) A Theoretical Model for Template-Free Synthesis of Long DNA Sequence. Syst. Synth. Biol. 2:67-73
| + | <li>Minhaz Ud-Dean, S.M. (2008) A Theoretical Model for Template-Free Synthesis of Long DNA Sequence. Syst. Synth. Biol. 2:67-73</li> |
| - | <BR> | + | <li>Koukhareva, I. and Lebedev, A. (2009) 3'-Protected 2'-Deoxynucleoside 5'-Triphosphates as a Novel Tool for Heat-Triggered Activation of PCR. Anal Chem. 81(12):4955-4962</li> |
| - | 2.Koukhareva, I. and Lebedev, A. (2009) 3'-Protected 2'-Deoxynucleoside 5'-Triphosphates as a Novel Tool for Heat-Triggered Activation of PCR. Anal Chem. 81(12):4955-4962
| + | <li>Kuan, W.L., Joy, J. Mee, N.F., Perlyn, K.Z., Wen, T.S., Nguen, T., James, J., Chai, E., Flotow, H., Crasta, S., Chua, K., Peng, N.S. and Hill, J. (2010) Generation of Active Bovine Terminal Deoxynucleotidyl Transferase (TdT) in E. coli. Biochemistry Insights. 3: 41-46.</li> |
| - | <BR> | + | <li>Boule, J.B., Rougeon, F.Papanicolaou, C. (2001) Terminal Deoxynucleotidyl Transferase Indiscriminately Incorporates Ribonucleotides and Deoxyribonucleotides. J. Biol. Chem. 276, 33: 31388-31393.</li> |
| - | 3.Kuan, W.L., Joy, J. Mee, N.F., Perlyn, K.Z., Wen, T.S., Nguen, T., James, J., Chai, E., Flotow, H., Crasta, S., Chua, K., Peng, N.S. and Hill, J. (2010) Generation of Active Bovine Terminal Deoxynucleotidyl Transferase (TdT) in E. coli. Biochemistry Insights. 3: 41-46.
| + | <li>Motea, E.A. and Berdis, A.J. (2010) Terminal Deoxynucleotidyl Transferase: The Story of a Misguided DNA Polymerase. Biochim. Biophys. Acta. 1804, 5: 1151-1166.</li> |
| - | <BR> | + | <li>Gardner, A.F., Wang, J., Wu, W., Karouby, J., Li, H., Stupi, B.P., Jack, W.E., Hersh, M.N. and Metzker, M.L. (2012) Rapid Incorporation Kinetics and Improved Fidelity of a Novel Class of 3'-OH Unblocked Reversible Terminators. Nucleic Acids Research. 40, 15: 7404-7415.</li> |
| - | 4.Boule, J.B., Rougeon, F.Papanicolaou, C. (2001) Terminal Deoxynucleotidyl Transferase Indiscriminately Incorporates Ribonucleotides and Deoxyribonucleotides. J. Biol. Chem. 276, 33: 31388-31393.
| + | <li>Kruszewski, Marcin, Elzbieta Bouzyk, Tomasz Oldak, Krystyna Samochocka, Leon Fuks, Wlodzimierz Lewandowski, Izabela Fokt, and Waldemar Priebe. "Differential Toxic Effect of Cis-platinum(II) and Palladium(II) Chlorides Complexed with Methyl 3,4-diamine-2,3,4,6-tetradeoxy-α-L-lyxo-hexopyranoside in Mouse Lymphoma Cell Lines Differing in DSB and NER Repair Ability." Teratogenesis, Carcinogenesis, and Mutagenesis 23.S1 (2003): 1-11. Web. 17 Oct. 2014.</li> |
| - | <BR> | + | <li>Hayakawa, Yoshihiro. "ChemInform Abstract: Toward an Ideal Synthesis of Oligonucleotides: Development of a Novel Phosphoramidite Method with High Capability." ChemInform 33.2 (2002)</li> |
| - | 5.Motea, E.A. and Berdis, A.J. (2010) Terminal Deoxynucleotidyl Transferase: The Story of a Misguided DNA Polymerase. Biochim. Biophys. Acta. 1804, 5: 1151-1166.
| + | <li>Palladium - Pd." Palladium (Pd). N.p., n.d. Web. 17 Oct. 2014.</li> |
| - | <BR> | + | <li>"Palladium and Its Compounds Toxicity Reports, Review - Hazard Potential, Risk." N.p., n.d. Web. 17 Oct. 2014.</li> |
| - | | + | |
| - | | + | |
| - | | + | |
| | | | |
| | | | |
| Line 123: |
Line 159: |
| | xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx --> | | xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx --> |
| | </div> | | </div> |
| | + | |
| | <br> | | <br> |
| | <br> | | <br> |
| Line 131: |
Line 168: |
| | | | |
| | | | |
| - |
| |
| - | <div class="footer">
| |
| - |
| |
| - | <div class="footer-blurb">
| |
| - |
| |
| - |
| |
| - | <br><br>
| |
| | | | |
| | | | |
| | + | <div class="cu-footer"> |
| | + | <div class="bottom-left"> |
| | + | <a href="https://2014.igem.org/Team:Cooper_Union" span="The Cooper Union: μToolbox"><img src="https://static.igem.org/mediawiki/2014/b/bd/CU_2014_logo.png" height="150" /></a></div> |
| | + | <div class="bottom-center"> |
| | + | <a href="https://2014.igem.org/Team:Cooper_Union/Project" class="black" > |
| | + | <img src="https://static.igem.org/mediawiki/2014/2/27/CU_2014_LogoP.png" height="100" style="vertical-align: top;" title="Projects" /> |
| | + | <span>Projects</span> |
| | + | </a> |
| | + | <a href="https://2014.igem.org/Team:Cooper_Union/Social" class="red" > |
| | + | <img src="https://static.igem.org/mediawiki/2014/b/b2/CU_2014_logoS.png" height="100" style="vertical-align: top;" title="Social" /> |
| | + | <span>Social</span> |
| | + | </a> |
| | + | <a href="https://2014.igem.org/Team:Cooper_Union/Notebook" class="green" > |
| | + | <img src="https://static.igem.org/mediawiki/2014/d/da/CU_2014_LogoN.png" height="100" style="vertical-align: top;" title="Notebook" /> |
| | + | <span>Notebook</span> |
| | + | </a> |
| | + | <a href="https://2014.igem.org/Team:Cooper_Union/biofanfic"class="blue"> |
| | + | <img src="https://static.igem.org/mediawiki/2014/3/38/CU_2014_LogoT.png" height="100" style="vertical-align: top;" title="Team" /> |
| | + | <span>Team</span> |
| | + | </a> |
| | + | </div> |
| | + | <div class="bottom-right"> |
| | + | <a href="http://www.cooper.edu" target="_blank" span="The Cooper Union"><img src="https://static.igem.org/mediawiki/2014/9/90/CU_2014_Cooper_union_logo.png" height="150" /></a> |
| | + | </div> |
| | </div> | | </div> |
| | | | |
| - |
| |
| - |
| |
| - | <div class="footer-links">
| |
| - |
| |
| - | <br><br>
| |
| - |
| |
| - |
| |
| - |
| |
| - | </div>
| |
| - |
| |
| - |
| |
| - | <div class="sponsors">
| |
| - |
| |
| - |
| |
| - | <br><br>
| |
| - |
| |
| - |
| |
| - |
| |
| - | </div>
| |
| - |
| |
| - |
| |
| - |
| |
| - | <br class="clear-left" />
| |
| - | </div>
| |
| | | | |
| | | | |
De novo Enzyme Mediated Oligonucleotide Synthesizer
De novo synthesis is a way of creating DNA oligonucleotides without the need of a template strand. Since the conventional method is expensive, time consuming, and inefficient, our team has focused on minimizing the time and money required for DNA synthesis while allowing labs to produce oligonucleotides easily without ordering. Here we introduce the De novo Enzyme Mediated Oligonucleotide Synthesizer (DEMOS); a programmable enzyme based, template-free, synthesizer for nucleic acid polymers. Our eventual goal will be to build a microfluidic De novo synthesizer that will allow laboratories, from academia to DIYBio community labs to rapidly and economically synthesize any strand of DNA for their projects. We hope that this system will become the key platform that bridges the
in silico to
in vitro gap in the design-test-build cycle of DNA synthesis and experimentation.
Our system is fundamentally based on two key technologies, one practical and the other theoretical:
- The development of deoxynucleotide triphosphate (dNTP) substrates with 3' reversible protective groups for "sequencing by synthesis" and "hot start" PCR technologies
- The published theoretical model of directed template-free synthesis of DNA using the enzyme terminal deoxynucleotidyl transferase (TdT). This enzyme that has the ability to add nucleotides to the 3' ends of DNA, preferable 3' overhangs, in a template-free manner.
In the general scheme shown below in Figure 1, an incoming 3'-RPdNTP (reverse protective group dNTP) is added to a free 3'OH of a growing DNA chain by the enzyme TdT at 37° Celsius. This is followed by a wash step where unincorporated 3'RPdNTPs and PPi (pyrophosphate) are removed. For heat labile 3'-RPdNTP’s deprotection is achieved by raising the temperature to 95° Celsius, while photolabile 3'-RPdNTP’s are deprotected by pulses of ultra-violet light. This is followed by another wash step where the decoupled protective groups are removed, thereby resetting the system to begin another cycle of addition.
 Figure 1. General DNA De novo Synthesis Scheme
Figure 1. General DNA De novo Synthesis Scheme
The deprotection reaction of the 3'-RPdNTP’s is shown in Figure 2a. The structures of two heat labile 3' protective group dNTPs that are commercially available from TriLink Biotechnologies (www.trilinkbiotech.com) are shown in Figure 2b . For our proof of concept experiments, 3'-TBE-dNTP’s were used, as it has a faster rate of deprotection at 95° Celsius; 3'-TBE-dNTP t 1/2 + 5 minutes, 3'-THF-dNTP t 1/2= 90 minutes)
 Figure 2 a. General Deprotection of Protected Nucleotide b. Thermolabile Protective Groups
Figure 2 a. General Deprotection of Protected Nucleotide b. Thermolabile Protective Groups
Advantages of Our System vs. Phosphoramidite Mediated Oligonucleotide Synthesis
 Figure 3. General Phosphoramidite Synthesis Cycle Scheme
Figure 3. General Phosphoramidite Synthesis Cycle Scheme
Overall, there are several notable advantages of our system over the Phosphoramidite system. Our system is substantially more eco-friendly, as some of the reagents used in the Phosphoramidite system are considered toxic. For instance, Palladium (Pd) complex is used to remove the protecting group from the dNTPs. Pd is considered to have low toxicity because the absorbency rate in the body is relatively low. However when the Pd complex is used as a reagent, it is possible for its toxicity to increase. There have been cases where it has caused acute toxic effects in lab mice, including mutagenesis to their isolated hearts. Another toxic reagent used is Dichloroacetic acid (DCA). DCA has been listed by the Environmental Protection Agency as a cancer-causing agent in humans and is also known to cause infertility in men. Additionally, long term use is associated with increased risk of liver cancer. Thus the use of these reagents is not cost effective since many precautions must be taken for their use. Permits must be obtained for their use, proper lab and safety equipment must be installed, safety protocols must be followed, and the reagents must be disposed of properly. Another advantage our system is that it is faster overall. As shown above in Figure 3, one cycle for the Phosphoramadite system is comprised of four main steps, while ours has only two. This greatly increases the kinetics of the system since the time it takes to complete a full synthesis is equivalent to the time that it takes to complete one cycle multiplied by the number of base pairs that will be synthesized.
Advantages of Our System vs. pH Regulated De novo Enzyme Mediated DNA Synthesis
In the controllable system proposed by Ud-Dean, the reversibility of a 3' acetyl protective group was achieved by lowering pH during each cycle and thereby activating a deacetylase enzyme that is active at lower pH. During this step, TdT is also inactivated by the shift to lower pH, thereby preventing the addition of extra nucleotides.
 Figure 4. General pH Regulated De novo Enzyme Mediated DNA Synthesis Cycle Scheme
Figure 4. General pH Regulated De novo Enzyme Mediated DNA Synthesis Cycle Scheme
Our system simplifies this novel approach by using thermolabile reversible 3' protective groups. Since our system uses heat to deprotect the dNTPs, only one step is needed instead of two, decreasing the time that is needed to add one base pair. This greatly increases the kinetics of the system since the time it takes to complete a full synthesis is equivalent to the time that it takes to complete one cycle multiplied by the number of base pairs that will be synthesized.
Results
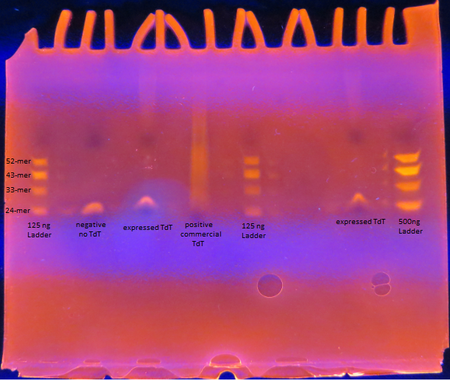 Figure 5. TdT Enzyme Functional Assay
Figure 5. TdT Enzyme Functional AssayAs seen from the polyacrylamide gel shown above in Figure 5, the TdT enzyme expressed in the Rosetta cells using our genetically engineered plasmid works as intended; base pairs are appended to oligos by the enzyme. However, there is a difference between the activity of our enzyme compared to the commercially obtained TdT. The majority of the oligos have only a few base pairs added by our expressed TdT while the lane of the commercial TdT is a smear of oligos, suggesting the commercial TdT has a greater reaction rate. But it can also be attributed to the fact that the 0.5 U/μL concentration of our expressed TdT is an overestimation. The TdT concentration of the reaction with our expressed TdT is less than that of the commercial TdT.
There is also a large gap between the oligos with only a few bases added and those with 30+ bases added. This suggests that our expressed TdT has a higher processivity than that of commercially obtained TdT. This might be due to subtle sequence differences such as the His tag on our TdT sequence.
Potential Errors in the Synthesis System
During each cycle there are independent sources of error that can affect the overall efficiency of the reaction. The most obvious errors include:
- Incomplete enzymatic addition of 3' reversibly protected dNTPs by TdT during the nucleotide incorporation step.
- Incomplete hydrolysis of 3' protective group during heating or other treatment.
- Incomplete removal of unincorporated 3' reversibly protected nucleotides after each incorporation step.
- Spontaneous hydrolysis of 3' protective groups prior to intended deprotection. Each error, if not sufficiently addressed would compound the likelihood of misincorporated nucleotides and a reduction of overall yield.
For example, during error scenario 1, oligonucletide chains that have not had a 3' reversibly protective group nucleotide added during an incorporation step, will, during the subsequent step after deprotection, be potential substrates for the addition of the next incoming nucleotide. This would lead to the base deletion. During error scenario 2, the failure to remove a protective group would lead to a population of oligonucleotides that would fail to add a nucleotide during the next subsequent addition of incoming nucleotides, leading to a base deletion at this step. For error scenario 3, the presence of unremoved, unreacted nucletides would lead to misincorporation and hence base substitutions during each subsequent stage of oligonucleotide elongation. During Scenario 4, the presence of dNTPs without 3' protective groups would lead to the incorporation of multiple dNTPs, resulting in homopolymeric stretches of nucleotides in the growing oligonucleotide chain. This would lead to insertion mutations. For a truly reliable system to be built, these and potentially other sources of error will need to be adequately countered.
Future Improvements to the De novo Oligonucleotide Synthesizer
In our present configuration, full length recombinant bovine TdT was used in the addition of thermolabile dNTPs to single stranded oligos. This particular enzyme denatures under elevated temperatures. Therefore, after the blocking group is removed from the dNTP at 95°C, it is necessary to add more of the enzyme. As a result, it would be useful to either isolate a naturally occurring variant of TdT or a similar enzyme that is resistant to denaturation under high temperature conditions. This type of enzyme might either be found in a thermophilic organism or synthetically engineered in a laboratory. Another potential improvement would be to construct truncated and other variants of engineered TdT that would incorporate modified dNTPs at a faster rate. Lastly, our current system could be improved by switching from commercially available thermolabile nucleotides to photolabile nucleotides. This should improve the system since temperature ramp up and down cycles are avoided, creating a faster reaction cycle and eliminating the need to re-add the TdT enzyme at the beginning of each cycle. We are presently exploring these improved features.
References
- Minhaz Ud-Dean, S.M. (2008) A Theoretical Model for Template-Free Synthesis of Long DNA Sequence. Syst. Synth. Biol. 2:67-73
- Koukhareva, I. and Lebedev, A. (2009) 3'-Protected 2'-Deoxynucleoside 5'-Triphosphates as a Novel Tool for Heat-Triggered Activation of PCR. Anal Chem. 81(12):4955-4962
- Kuan, W.L., Joy, J. Mee, N.F., Perlyn, K.Z., Wen, T.S., Nguen, T., James, J., Chai, E., Flotow, H., Crasta, S., Chua, K., Peng, N.S. and Hill, J. (2010) Generation of Active Bovine Terminal Deoxynucleotidyl Transferase (TdT) in E. coli. Biochemistry Insights. 3: 41-46.
- Boule, J.B., Rougeon, F.Papanicolaou, C. (2001) Terminal Deoxynucleotidyl Transferase Indiscriminately Incorporates Ribonucleotides and Deoxyribonucleotides. J. Biol. Chem. 276, 33: 31388-31393.
- Motea, E.A. and Berdis, A.J. (2010) Terminal Deoxynucleotidyl Transferase: The Story of a Misguided DNA Polymerase. Biochim. Biophys. Acta. 1804, 5: 1151-1166.
- Gardner, A.F., Wang, J., Wu, W., Karouby, J., Li, H., Stupi, B.P., Jack, W.E., Hersh, M.N. and Metzker, M.L. (2012) Rapid Incorporation Kinetics and Improved Fidelity of a Novel Class of 3'-OH Unblocked Reversible Terminators. Nucleic Acids Research. 40, 15: 7404-7415.
- Kruszewski, Marcin, Elzbieta Bouzyk, Tomasz Oldak, Krystyna Samochocka, Leon Fuks, Wlodzimierz Lewandowski, Izabela Fokt, and Waldemar Priebe. "Differential Toxic Effect of Cis-platinum(II) and Palladium(II) Chlorides Complexed with Methyl 3,4-diamine-2,3,4,6-tetradeoxy-α-L-lyxo-hexopyranoside in Mouse Lymphoma Cell Lines Differing in DSB and NER Repair Ability." Teratogenesis, Carcinogenesis, and Mutagenesis 23.S1 (2003): 1-11. Web. 17 Oct. 2014.
- Hayakawa, Yoshihiro. "ChemInform Abstract: Toward an Ideal Synthesis of Oligonucleotides: Development of a Novel Phosphoramidite Method with High Capability." ChemInform 33.2 (2002)
- Palladium - Pd." Palladium (Pd). N.p., n.d. Web. 17 Oct. 2014.
- "Palladium and Its Compounds Toxicity Reports, Review - Hazard Potential, Risk." N.p., n.d. Web. 17 Oct. 2014.
 "
"




