 "
"
Team:Evry/Notebook
From 2014.igem.org
| Line 78: | Line 78: | ||
<div id="test123"> | <div id="test123"> | ||
</div> | </div> | ||
| + | |||
| + | |||
<script language="JavaScript"> | <script language="JavaScript"> | ||
| Line 169: | Line 171: | ||
</div>--> | </div>--> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| + | </div> | ||
| Line 174: | Line 180: | ||
| + | <section id="speakers"> | ||
| + | <!-- <div class="col-lg-10 col-lg-offset-1"> --> | ||
| + | <div id="st-container" class="st-container"> | ||
| - | + | <h5>RNAseq</h5> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</html> | </html> | ||
Revision as of 15:43, 18 August 2014
Notebook
RNAseq
Week 1
Here
Contents |
Transformation
Week 1
Week 1
HERE HERE
Sensing
Notebook - Sensing

Mutation of the Pst1 site
Digestion of plasmid containing DmpR + GFP on three strains of E.coli (DH5a, Top10 and Bl21) by EcoR1 (wells 1), Pst1 (wells 2) and EcoR1+Pst1 (wells 3).

We have the good construction in DH5a AND tOP 10 STRAINS.
Assembling of the parts
Ligation of all parts
Transformation on E.coli
Purification of palsmids with NucleoSpin Plamsid Kit (Macherey Nagel) on 4 colonies
Digestion with EcoRI

All colonies contain the construction of all parts.

Sensor construction bphR2/PbpR1
A new tentative of golden gate has been done with modification of volumes of each part:
- J23114-rbs : 0,12µL
- bphR2: 1,05µl
- bphR1:0,31µl
- RFP : 0,64µl
- Terminator: 0,67µl
- pSB1C3,G3: 1µl
The mix enzyme was inchanged and the program is of 8h
Sep 08
Sensor construction bphR2/PbpR1
Results PCR clean-up:
- J23114-rbs : 76ng/µl
- bphR2: 76,8 ng/µl
- bphR1:126,4 ng/µl
- RFP : 98,3ng/µl
- Terminator: 42,4ng/µl
- pSB1C3,G3: 1µl
Sep 07
Sensor construction bphR2/PbpR1
Miniprep was made for each part and the nanodrop gives:
- J23114-rbs: 205 ng/µl
- bphR2: 227,5ng/µl
- bphR1:57,2ng/µl
- RFP: 74,2ng/µl
- terminator:54,6ng/µl
- pSB1C3,G3: 182,7ng/µl
A new PCR has been done by using Q5 DNA polymerase for all the parts expected for the terminator (one taq polymerase)
Sep 05
Sensor construction bphR2/PbpR1
As the first golden gate test failed, we retry at the beginning. Parts were cultured in 10ml LB + 10µL Cam for bphR2, bphR1, RFP, Terminator, pSB1C3 and in 10ml LB + 10µL Amp for J23114 and stored at 37°C overnight
Sep 04
Sensor construction bphR2/PbpR1
There is not colonies from the transformation of golden gate product in pSB1C3,G3.
Sep 03
Sensor construction bphR2/PbpR1
Transformation has made with golden gate product and 5µL of the product migrated :

There is'nt DNA in the mix
Aug 29
Sensor construction bphR2/PbpR1
PCR colony has been done with colonies of the transformation bphR2 in pSB1C3. The tail expected was 1242bp and we obtained it:

A nanodrop has been done on the colony 1 (48ng/µl) and colony 2 (4&,4ng/µl) and the colony has been send to sequencing
With PCR clean up of each part, golden gate has been done. Volumes for each part has been calculated with cloning bench application.
Mix DNA contains:
- J23114-RBS (82,1ng/µl):0,06 µl
- bphR2 (139,3 ng/µl) : 0,327µl
- bphR1 (101,1ng/µl): 0,21µl
- RFP (75,3 ng/µl): 0,46µl
- Terminator (28,1ng/µl) : 0,37µl
- pSB1C3,G3 (107,2ng/µl) : 0,93µl
Mix enzyme contains:- 1,5µl 10X T4 ligase buffer
- 0,5µl BSAI
- 0,5µl T4 ligase
- 2,5µl
Aug 28
Sensor construction bphR2/PbpR1
Some colonies had grown after the transformation of bphR2 in pSB1C3:

Products PCR from 26th august has been migrated :

Expected for the terminator, we have bands at the good tail. So for the terminator, an other PCR has been made but by using the one taq polymerase instead of Q5 DNA polymerase and we obtained the good band:

Aug 27
Sensor construction bphR2/PbpR1
We wanted to assemble the construction by using golden gate. After to have received primers, PCR was done to amplify J23114-RBS, bphR2, bphR1-RBS, RFP and terminator with golden gate primers. The tail of each part is:
- J23114-RBS = 82bp
- bphR2 mutated = 929bp
- bphR1-RBS = 340bp
- RFP = 732bp
- Terminator = 162bp
Simultaneously, the product of digestion of bphR2 and pSB1C3 has been migrated to verify the digestion then, bphR2 and pSB1C3 has been ligated and we have done a heat shock transformation.
Aug 26
Sensor construction bphR2/PbphR1 :
Miniprep of pSB1C3 was done => 68,9ng/µl and the plasmid was digested with EcoRI-HF and pstI.
Digestion products of pSB1C3 and bphr2 has been migrated on gel 1X agarosis. Bands expected were:
- for pSB1C3: one band at 2069bp (vector) and one band at 1070bp (insert)
- for bphr2: one band at 946 pb (insert) and one band at 1300bp (vector)
We obtained the good bands so an extraction on gel was done to recover only pSB1C3 without its insert and only bphr2 gene. Aug 23
Transformation plate observation:
- BBa_E1010: 50-60 colonies
- BBa_J23114: 150-200 colonies
- BBa_B0015: 40-50 colonies
- PSB1C3G3: > 1000 red colonies
A PCR was performed on 8 colonies for BBa_J23115 (K823012) following the protocol Table 3 and 4.
Samples are loaded on a 1% agarose gel, 10 µL of sample + 2 µl of loading dye 6X per well. Gel ran 45 minutes at 100 mV.
Sensor construction bphR2/PbphR1 :
One colony of the transformation of pSB1C3 was incubated in 3ml LB + 3µL Cam, at 37°C, overnight
Sensor construction bphR2/PbpR1
BBa_J23114 was resuspended with 10 µL sterile water in order to obtain a DNA concentration around 0.2 ng/µl (according to the registry). Solution was transferred into one 1 ml eppendorf tube and stored at -20°C.
Transformations of constitutive promoter BBa_J23114, RFP BBa_E1010 and terminator BBa_B0015 and vector pSB1C3G3 was done on DH5alpha as followed:
- Remove E. coli competent tubes from -80°C and keep it on ice
- Add 3 µl of template (here solubilized plasmids from the registry distribution kit) and mix gently
- Incubate 10 minutes on ice
- Perform an heat shock 30 seconds at 42°C
- Incubate 2 minutes on ice
- Add 2 ml of LB medium and incubate 60 minutes at 37°C with an agitation at 200 rpm
- Plate 200 µl of pSB1C3G3, BBa_B0015 and E1010 on a chloramphenicol LB agar plate and ampicilline LB agar plate for BBa_J23114
- Incubate plate overnight at 37°C
We received primers.

pSB1C3 was digested with EcoRI and pstI and a ligation with bphR2 was done before the transformation in DH5a.
50 µL were plated on Cam Lb plate and incubated at 37°C overnight
Aug 21
Sensor construction bphR2/PbphR1
We received sequencing reads: that match perfectly to the registry sequence.
26DJ54
AATAGGCGTTATCACGAGGCAGAANTTCAGATAAAAAAAATCCTTAGCTTTCGCTAAGGATGATTTCTGGAATTCGCGGCCGCTTCTAGAGATGTCCCTGG
GTGACATGCGTGATTTGGCCGCCACGCGGATCGCGCTCGAGAGCGAAGCGTTACGCCAAAGCGTGCTGAATGGTGACGCTGAATGGGAGGCGCGGATCGTCAGTTC
GTTTCACCGACTGTCATTGATTGAAGAGCCCACGATGCGGGATCCGGCTCGCTGGTTTAATGAGTGGGAGCCAGTCAACCGCGGTTTTCACGAAGCTCTTATCTCT
GCCTGTTCGTCCGTCTGGATCCGGCGGTTCCTGTCCATCCTGTATGTGCATATGGAGCGCTACCGCCGATTGACTGCTTACTAGTAGCGGCCGCTGCAGTCCGGCA
AAAAAGGGCAAGGTGTCACCACCCTGCCCTTTTTCTTTAAAACCGAAAAGATTACTTCGCGTTATGCAGGCTTCCTCGCTCACTGACTCGCTGCGCTCGGTCGTTC
GGCTGCGGCGAGCGGTATCA
GCTCACTCAGGG
26DJ55
CGAGTCAGTGAGCGAGGAAGCCTGCATAACGCGAAGTAATCTTTTCGGTTTTAAAGAAAAAGGGCAGGGTGGTGACACCTTGCCCTTTTTTGCCGGACTGC
AGCGGCCGCTACTAGTAAGCAGTCAATCGGCGGTAGCGCTCCATATGCACATACAGGATGGACAGGAACCGCCGGATCCAGACGGACGAACAGGCAGAGATAAGAG
CTTCGTGAAAACCGCGGTTGACTGGCTCCCACTCATTAAACCAGCGAGCCGGATCCCGCATCGTGGGCTCTTCAATCAATGACAGTCGGTGAAACGAACTGACGAT
CCGCGCCTCCCATTCAGCGTCACCATTCAGCACGCTTTGGCGTAACGCTTCGCTCTCGAGCGCGATCCGCGTGGCGGCCAAATCACGCATGTCACCCAGGGACATC
TCTAGAAGCGGCCGCGAATTCCAGAAATCATCCTTAGCGAAAGCTAAGGATTTTTTTTATCTGAAATTCTGCCTCGTGATACGCCTATTTTTATAGGTTAATGTCA
TGATAATAATGGTTTCTTAGA
Plasmid with bphR2 gene was purified with NucleoSpin Plamsid protocol (Macherey-Nagel) => we obtained 227,5ng/µL
This plasmid was digested according this protocol:
- Add:
- sterilized water: qsp 20µL
- template DNA : 500ng
- buffer 2.1: 2µL
- BSA: 0,2µL
- EcoRI: 1µL
- PstI: 1µL
- Reverse for mix
- Incubate at 37°C during 45mn
- Incubate at 80°C during 20mn
- Ligation was not possible because pSB1C3 plasmid was not digested so the digested plasmid (bphR2 gene) was stored at -20°C
Survival test on E.coli BL21:
Bacteria survive again for different concentrations but they were very concentrated
A dilution of the medium has been done for the positive control and the six different concentrations:

PCR using VF2 and VR primer
Q5 polymerase
Expected bands :
DmpR: 2038 bp
GFP B0031: 1331 bp
GFP B0032: 1330 bp
Digestion
From newly extracted DNA
DmpR: SpeI&PstI
GFP B0031/32: XbaI&PstI

VF2/VR PCR products are still in agreement with the expected size either for DmpR and GFP B0031/32.
GFP 31/32 digestion products were in agreement with the expected size.
DmpR digestion by SP is still displaying 2 close bands profile.
Aug 20
Sensor construction bphR2/PbphR1:
A colony after the DH5a transformation was cultured in 3mL of LB + 3µL Amp, at 37°C, 200rpm, overnight
Survival test on E.coli BL21:
Bacteria continue to grow for all concentrations of 2 hydroxy-3',4'dichlorobiphenyl.
Dna extraction
Machery Nagel DNA purification Kit (PROTOCOL)
PCR using VF2 and VR primer
Q5 polymerase
Expected bands :
DmpR: 2038 bp
GFP B0031: 1331 bp
GFP B0032: 1330 bp
Digestion
DmpR: SpeI&PstI
GFP B0031/32: XbaI&PstI


Analysis
VF2/VR PCR products were in agreement with the expected size either for DmpR and GFP B0031/32.
GFP 31/32 digestion products were in agreement with the expected size.
However DmpR digestion revealed an unexpected profile.
Gel extraction
Extraction of GFP B0031/32 digestion product [XbaI-PstI]
After confirmation of relevant electrophoresis bands, cut the gel all around the band and as close as possible to it.
Place the resulting piece into a 2ml microcentrifuge tube. Add 1µl of Binding buffer to 1 µg of gel. Place the tube at 55°C to 60°C during 10min or until gel turn completely liquid.
Follow classical DNA purification protocol.
Machery Nagel kit (PROTOCOL 2)
Sensor construction bphR2/PbphR1:
bphR2 gene, synthetized by Eurofins in a plasmid, was dissolved in 23,5µL of elution buffer because it was lyophilized.
Concentration obtained = 200ng/µL
DH5a chimiocompetents were transformed with this plasmid according to this protocol:
- Remove DH5a from -80°C (about 200µL) and let them on the ice
- Add 100ng of plasmid at DH5a and let during 30mn on ice
- Make a heat shock by putting bacteria at 42°C during 30s-1mn
- Let 1h at 37°C
- Plate 100µL of the culture on LB-agar-Amp
- Incubate at 37°C overnight, 200rpm
Aug 17

Cell culture of E.coli received from registry
BBa_K1031211 (Pr-DmpR )
BBa_K1031221 (P0-RBS B0031-GFP)
BBa_k1031222 (P0-RBS B0032-GFP)
Liquid : 3ml of Luria Broth 1x + 34µg/L Chloramphenicol in a 15ml falcon at 37°C shaking
On plate : 25ml LB 1x + 25ml
Agarose 1x + 34µg/L Chloramphenicol
Observation: DmpR bacteria have difficulties to grow compared with GFP bacteria

Left : BBa_K1031221 (P0-RBS B0031-GFP)
Right : BBa_k1031222 (P0-RBS B0032-GFP)
Survival test on E.coli BL21:
- Serial dilution of compound was made from 10(-2) mol/L to 10(-8) mol/L

- 300µL of E.coli BL21 were added in each eppendorf tube.
We had 2 control tubes:- A negative control which contains 2,7mL of M9 medium + 300µL of compound
- A positive control which contains 2,7mL of M9 medium + 300µL of E.coli BL21
- Tubes were incubated at 37°C overnight, 200rpm Aug 15
Survival test on E.coli BL21:
This test allowed to know if E.coli can survive at different concentrations of 2 hydroxy-3',4'dichlorobiphenyl
- For that, BL21 culture was removed from the -80°C and culturing in 4 mL of LB
- E.coli was incubated overnight at 37°C, 200rpm
Survival test on E.coli BL21:
10mg of 2 hydroxy,3'-4'dichlorobiphenyl have been received. The compound was lyophilized so it's has been dissolved in 17mL of DMSO in order to have a final concentration of 10(-2)M
The compound was stored at -20°C, in the dark because the DMSO is sensitive to the light.
Aug 12
Survivability in molecules which are going to be sensed - Results:
Here are the results of the survivability of Pseudovibrio in media with molecules which are going to be sensed:

Pseudovibrio seems to survive in presence of all concentrations of nitrite, it's affect cells' growth but don't kill Pseudovibrio.
In Lead, big concentrations lead to cells' death, but Pseudovibrio survive very well until a concentration of 5µL.
Cadmium does'nt seem to block cells' growth in concentrations which we want to use.
Survivability in molecules which are going to be sensed:
Ranges of concentrations of molecules that we want to sense were made as described in following tables:

1. Serial dilutions of compound were made



2. 300µL of E.coli BL21 was added in each eppendorf tube.
We had 2 control tubes:
*A negative control: 2,7mL of MB medium + 300µL of compound
*A positive control: 2,7mL of MB medium + 300µL of E.coli BL21
3. Incubation of tubes at 30°C overnight, 200rpm Jul 29
Sensor construction bphR2/PbphR1:
- Plate 20µL from the culture of 27th july on LB-agar-Cam (25 mL LB-Agar + 25µL Chloramphenicol)
- Incubation of plate at 37°C overnight
- A glycerol stock was made with 750ml of culture and 750ml of glycerol 50% and stored at -80°C
Sensor construction bphR2/PbphR1:
- Culturing of one colony bphR1 promoter (BBa_K1155001), received from Paris-Saclay's team, in 5ml LB + 5µL Chloramphenicol (Cam)
- Incubation at 37°C overnight, 200rpm
Interlab study
Week 7
Interlab Study
08.11.2014
08.12.2014
The 4 needed parts are: BBa_J23101, BBa_J23115, BBa_E0240 and BBa_I20260. Corresponding wells were located on 2014 Distribution kit plates and resuspended with 10 µL steril water. That permits to obtain a DNA concentration around 0.2 ng/µl (according to the registry. Solutions were transferred into 1 ml eppendorf tubes and stored at -20°C.
To amplify fragments, a PCR was performed on the 4 constructions, with the mix described on table 1.

Distribution of 49 µl of mix per PCR tube. Application of program IGEM Q5 PCR.

Preparation of a 1% agarose gel: 0.56 g of Top Vision agarose (Thermo Scientific) + 50 ml of TAE 1X.Microwave 30s by 30s until agarose total dissolution. Gel was cooling down until to be lukewarm, one BET drop was added. Gel was loaded with 10µl per sample previously added with 2 µl of loading dye 6X, and 5 µl for ladders. Gel running 45 minutes at 100 mV in TAE 1X buffer.

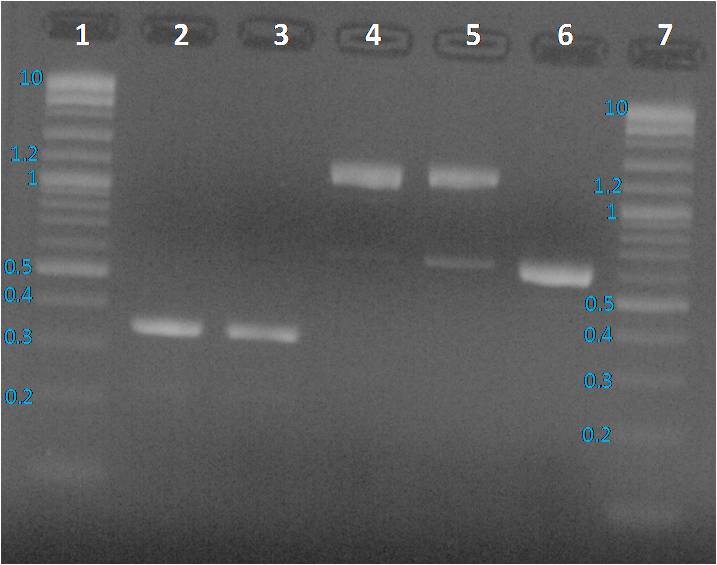
Lane 1: BBa_J23115 PCR product, Lane 2: pBHR1 PCR product, Lane 3 and 6: Purple 2-Log ladder NEB, Lane 4: BBa_E0240 PCR product, Lane 5: BBa_I20260 PCR product, Lane 7: 1 Kb plus Ladder Ogene Ruller and Lane 8: BBa_J23101 PCR product
We expected to obtain one band per PCR sample corresponding to the interesting amplified fragment. For Lane 2, 3 and 6 it was ok. We had the expected profile. By contrast 2 bands were visible, one at the expected size (around 1200 bp) and another around 600 bp. We decided to perform a purification on gel of each bands.
08.13.2014
PCR products were cleaned with the GeneJET purification kit (Thermo Scientific)followed by DNA quantification with the NanoDrop 2000 (Thermo Scientific).
Preparation of a 1% agarose gel: 0.56 g of Top Vision agarose (Thermo Scientific) + 50 ml of TAE 1X. Microwave 30s by 30s until agarose total dissolution Gel was cooling down until to be lukewarm, one BET drop was added. Gel was loaded with 10µl per sample previously added with 2 µl of loading dye 6X, and 5 µl for ladders. Gel running 45 minutes at 100 mV in TAE 1X buffer.
Profiles were same as before PCR clean up. To sequence the amplified parts, we decided to a purification of fragments from the gel as presented below.
Preparation of a 1% agarose gel: 0.56 g of Top Vision agarose (Thermo Scientific) + 50 ml of TAE 1X. Microwave 30s by 30s until agarose total dissolution. Gel was cooling down until to be lukewarm, one BET drop was added. Gel was loaded with 30 µl of BBa_E0240 purified PCR product and BBa_I20260 purified PCR product added with 6 µl of loading dye, at 10µl of the mix per well. Gel running 45 minutes at 100 mV in TAE 1X buffer.

Four pieces of gel were sampled in four tubes to perform a DNA purification from gel.
08.14.2014
To amplify BBa_I20260 and BBa_0240, a PCR was perform with the mix described Table 1. The program IGEM Q5 PCR was applied, see Table 2. PCR products were cleaned with the GeneJET purification kit (Thermo Scientific)followed by DNA quantification with the NanoDrop 2000 (Thermo Scientific).
Preparation of a 1% agarose gel: 0.52 g of Top Vision agarose (Thermo Scientific) + 50 ml of TAE 1X. Microwave 30s by 30s until agarose total dissolution Gel was cooling down until to be lukewarm, one BET drop was added. Gel was loaded with 20 µl of BBa_E0240 PCR product and BBa_I20260 purified PCR product added with 4 µl of loading dye, at 10µl of the mix per well.
Gel running 45 minutes at 100 mV in TAE 1X buffer.

3 pieces of gel were sampled in four tubes to perform a DNA purification from gel. The DNA purification was made with the GeneJET purification kit (Thermo Scientific.
A verification electrophoresis was performed on a 1% agarose gel.

The major part of the DNA was lost during the purification because the amount of was to weak to be purified from gel. So we decide do transform ''E. coli'' to isolate a colony containing the desired plasmid for BBa_E0240 and BBa_I20260.
08.15.2014
BBa_E0240 and BBa_I20260 are respectively into a PSB1C3 and a PSB1K3 vector. In order to select colonies, LB complemented with kanamycin or chloramphenicol are necessary. 200 ml of LB agar medium was prepared with 7 g of LB Agar powder(Sigma) into 200 ml of miliQ water. After complete dissolution, the solution was sterilized in the Tuttmauer 2540ML apparatus, STE 20 minutes at 121°C and EXT+DRY 15 minutes. 50 µl of kanamycin stock solution (25 mg/L) was added to 50 ml of LB agar to pour 2 plates. 150 µl of chloramphenicol stock solution was added in the 150 LB agar remaining ml to pour 6 plates.
The transformation was performed on DH5 alpha ''E. coli'', as followed:
- Remove E. coli competent tubes from -80°C and keep it on ice
- Add 1 µl of template (here solubilized plasmids from the registry distribution kit) and mix gently
- Incubate 10 minutes on ice
- Perform an heat shock 30 seconds at 42°C
- Incubate 2 minutes on ice
- Add 3 ml of LB medium and incubate 60 minutes at 37°C with an agitation at 200 rpm
- Plate 200 µl of BBa_E0240 on a chloramphenicol LB agar plate and BBa_I20260 on a Knamycin LB agar plate
- Incubate plate overnight at 37°C
08.16.2014
Nothing had grown during the night on both plates. Taking into consideration different hypothesis, the transformation was repeated with a change on step 6 and 7. At step 6, only 1 ml of LB medium was added. To plate, 200 µl of BBa_E0240 and BBa_I20260 were plated on 3 plates: LB agar, LB agar kanamycin and LB agar chloramphenicol.
08.17.2014
Colonies had grown on plates as followed:- LB agar: more than 1000 colonies for the two biobricks
- LB agar kanamycin: 10 colonies for BBa_E0240 and >40 colonies for BBa_I20260
- LB agar chloramphenicol: >50 colonies for BBa_E0240 and 3 colonies for BBa_I20260
Plate storage at 4°C.
Cell Characterization
Notebook - Cell Characterization

New protocols of electrocompetent Pseudovibrio - Results

The transformation didn't work with Pseudovibrio washed 5 times or 6 times.
New protocols of electrocompetent Pseudovibrio
Transformation was perfomed on the new stocks made the 10-06-14 with two types of washes.
1µL of plasmids PBBR1MCS and pRhokHI-2 (25 and 33ng/mL).
2000 V
(Protocole)
New protocols of electrocompetent Pseudovibrio
On a pre-culture of Pseudovibrio in MB 1X 3mL, we relaunch the culture in 100mL of MB 1X. We made the electrocompetent protocol (Protocole).
We tested to make 5 and 6 washes with glycerol 10% instead of three.
Stocks of cells were stored at -80°C (3 weeks maximum)
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Confirmation
We tried to confirm the presence of our plasmid of previous transformations by extracting potential plasmids and digest them with EcoRI. After the incubation and the inactivation of the enzyme, products were migrating.
No band on the gel. The control (lab stock) didn't work either, so there is probably a problem of quantity of DNA.
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Results
The transformation didn't work.


Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Confirmation
A contamination has been shown in another transformation test. We can't use our previous results because there is a chance that it's not Pseudovibrio but the contamination which had been transformed.
We can still try to prove that it's our bacteria (with amplification of specific sequence) ond that there is our plasmids.
New transformation was tried with another stock of competent Pseudovibrio (Protocole) and cells were plated on MB/MB+Cam(1:1000)/MB+Kan(1:1000).
Stock of pBBR1MCS and pRhokHI-2
The colony of E.Coli transformed in LB 1X + Cam (1:1000) culture of 3mL was sowed on new LB/LB+Cam(1:1000)/LB+Kan(1:1000) plates and incubated with shaking overnight at 37°C.
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Confirmation
The previous digestion could fail because of the too little time of incubation (15min). We re-try to make the digestion with an incubation of 1 hour.

There was a problem with the digestion. We are going to try with another enzyme.
Stock of pBBR1MCS and pRhokHI-2
To remake a stock of these plasmids, we showed a colony of E.Coli transformed in a new LB 1X + Cam (1:1000) culture of 3mL.
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Confirmation
We made a restriction control of our plasmid, and we tried to prove that these plasmids are present in our Pseudovibrio.
After made a purification of plasmids in A1 and A2 with NucleoSpin Plamsid Kit (Macherey Nagel), we obtained concentrations at 380.1 ng/µL for pRhokHI-2 and at 91.1 ng/µL for pBBR1MCS.
We disgested these sample and our stock of pRhokHI-2 and pBBR1MCS with XbaI (unique restriction site on these plasmids) and we have make migrate the product of digestion.
There was a problem with the digestion. We are going to try with a longer time of incubation.
Stock of pBBR1MCS and pRhokHI-2
To remake a stock of these plasmids, we put in liquid LB+Cam (1:1000) culture, a colony from a LB+Cam plate of E.Coli transformed.
New stock of electro competents Pseudovribio - Antibiotics' control

New stock of electro competents Pseudovribio
We choose to make a new stock of electro competent Pseudovibrio denitrificans.
We made a culture of 100mL from a pre-culture of 10mL made the 04/09.
The new stock was test on plates MB 1X with Cam and Kan (1:1000).
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Results


NB: The negative control with E.Coli does'nt work on any plates. The culture was probably too old.
A problem occurs with stocks of electro-competent Pseudovibrio because wa can see that it growth on Kanamycine 1:1000. On Chloramphenicol, the contamination does'nt seem to grow.
We have to prove that it's Pseudovibrio wich growth on Chloramphenicol. Sep 02
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Transformation
| pBBR1MCS | pRhokHI-2 | Ø plasmid |
| Pseudo replicate 1 | Pseudo replicate 1 | Pseudo |
| Pseudo replicate 2 | Pseudo replicate 2 | E.Coli Bl21 |
| E.Coli Bl21 | E.Coli Bl21 |
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Selection
Plates are showed and organised like shown in the following picture:

| A1 | B1 | C1 | D1 |
| Pseudo+pBBR1MCS (1) | Pseudo+pBBR1MCS (2) | Pseudo | Pseudo Ø plasmid |
| A2 | B2 | C2 | D2 |
| Pseudo+pRhokHI-2 (1) | Pseudo+pRhokHI-2 (2) | Pseudo | Pseudo Ø plasmid |
| A3 | B3 | C3 |
| E.Coli Bl21 | Bl21 Ø plasmid | Bl21+pRhokHI-2 |
| A4 | B4 | C4 |
| E.Coli Bl21 | Bl21 Ø plasmid | Bl21+pBBR1MCS |
We also made MB only plates to see the normal growth of our bacteria,which were showed and organised like shown in the following picture:

| A | B | C | D | a | b | c | d | e | f |
| E.Coli Bl21 | Bl21 Ø plasmid | Bl21+pBBR1MCS | Bl21+pRhokHI-2 | Pseudo | Pseudo Ø plasmid | Pseudo+pRhokHI-2 (1) | Pseudo+pRhokHI-2 (2) | Pseudo+pBBR1MCS (1) | Pseudo+pBBR1MCS (2) |
Sep 01
Transformation of Pseudovrbrio with pBBR1MCS and pRhokHI-2 - Competents cells
New stock of electro-competent Pseudovibrio was made from a pre-culture of 100mL.
(Protocole)
Amplification of pRhokHI-2 in E.Coli Bl21 - Results
After incubation overnight we can see some colonies of E.Coli Bl21 transformed on LB+Cam 1:1000 and on LB+Kan 1:1000. Our Bl21 which had not been transformed doesn't growth on these media but both grow on LB only.
We had well success to tranform E.Coli and amplifly the plasmid.

We tranfered two colonies into liquid MB+Kan(1:1000) and let them in incubator overnight to have a pre-culture for the plasmid purification with NucleoSpin Plamsid Kit (Macherey Nagel).
Aug 28
Differed launch of antibiotics' tests




Amplification of pRhokHI-2 in E.Coli Bl21
The transformation of E.Coli with pRhokHi-2 still didn't works. We still have to amplify it so we re-tried to the tranformation (1µL of plasmid for 40µL of cells, 1800V).
This time, after a liquid culture of only one hours cells were plated on LB 1X + Kan (1:1000), LB 1X + Cam (1:1000), and LB only and let in incubator overnight.
Differed launch of antibiotics' tests



Transformation of Pseudovribrio with pBBR1MCS - Results
We saw no colony on our plates. We decided to test our two replicates of transformation on MB 1X plates with other concentrations of antibiotic. We made three plates with Cam 1:5000, and 1:10 000, then Pseudovibrio were showed on them and let overnight in incubator.
Differed launch of antibiotics' tests



No readable results
Amplification of pRhokHI-2 in E.Coli Bl21
We re-tried to transform E.Coli with pRhokHI-2 to amplify it (1µL of plasmid for 40µL of cells, 1800V). This time, after a liquid culture of only one hours cells were plated on LB 1X + Kan (1:1000) and let in incubator overnight.
Transformation of Pseudovribrio with pBBR1MCS
After made a purification of pBBR1MCS with NucleoSpin Plamsid Kit (Macherey Nagel), electrocompetent Pseudovibrio cells were tranformed with the plasmid by electroporation (1µL of plasmid for 40µL of cells, 1800V), then they were incubated for three hours and showed on MB 1X+Cam (1:1000) plates. Plates are let in incubator overnight.
Differed launch of antibiotics' tests


Aug 25
Amplification of plasmids pBBR1MCS and pRhokHI-2 in E.Coli Bl21 - Results

Amplification of pBBR1MCS works and we purified it with NucleoSpin Plamsid Kit (Macherey Nagel) .
Unfortunately the transformation with pRhokHI-2 didn't works so we have to retry.
Differed launch of antibiotics' tests


Aug 24
Amplification of plasmids pBBR1MCS and pRhokHI-2 in E.Coli Bl21
Two plasmids pBBR1MCS and pRhokHI-2 were generously sent by Dr Thomas DREPPER (University Duesseldorf, Dept. of Biology, Institute of Molecular Enzyme Technology)

Bowth were transfered into Bl21 electro-competent cells by electroporation (two replicate)(1µL of plasmid for 40µL of cells, 1800V).
After a liquid culture of three hours, cells were plated on LB 1X + Cam (1:1000) and let in incubator overnight.
Antibiotics' tests on MB 1X plates - Results Day3
Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case.
Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.





Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case. Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.






Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case.
Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.






Differed launch of antibiotics' tests

Antibiotics' tests in liquid M9 for E.Coli transformed with a plasmid PSB1A3 containing an ampicilin resistance cassette were launch. In two days, these cultures will be plated on MB 1X, MB 0.5X and M9 1X plates
Aug 23
Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case.
Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.







Antibiotics' tests on MB 0.5X plates - Results Day2
Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case.
Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.







Antibiotics' tests on M9 1X plates - Results Day2
Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case. Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.





 Aug 22
Aug 22
Antibiotics' tests in liquid M9 1X - Results Day3






Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case. Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.






Antibiotics' tests on MB 0.5X plates - Results Day1
Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case. Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.






 "
"Antibiotics' tests on M9 1X plates - Results Day1
Results of antibiotics' tests on plate are calculated as percentage of bacteria's growth by compared with the controle case. Pseudovibrio coming from culture with antibiotics (part B on plates), are taken from antibiotics' range in M9 at Day2.
Antibiotics' tests in liquid M9 1X - Results Day2






Plates are showed and organised like shown in the following picture, and showed with 1µL of each culture:

| A | B | C | D |
| Pseudovibrio denitrificans | Pseudovribrio denitrificans* | E. Coli | E. Coli** |
**Bacteria tranformed with a plasmid containing a resistance cassette for the antibiotic
Aug 20
Antibiotics' tests in liquid M9 1X - Results Day1
- Chloramphenicol

- Kanamycin
 Because of technical problems, the tests on E.Coli+KanR are launched with a gap of few days.
Because of technical problems, the tests on E.Coli+KanR are launched with a gap of few days.
- Ampicilin

- Tetracyclin

- Erythromycin
 Aug 19
Aug 19
Tests of antibiotics' stocks - Results(2)
| Plates | LB Agar only | LB+Kan | LB+Erm (1:100) |
| E.coli | X | 0 | 0 |
New stocks are ready for antibiotics' tests on Pseudovribrio
Launch of antibiotics's tests in liquid M9 1X
For each tested antibiotic we made the following range of concentrations:
| 0 | 1:500 | 1:1000 | 1:50 000 | 1:100 000 |
We made these range two time to have a replicate and we added a controle tube without antibiotic for each bacteria.
Each tube is made like in the following table in Falcon tubes (15mL):
| / | 0 | 1:500 | 1:1000 | 1:50 000 | 1:100 000 |
| Medium+agar | 3mL | 3mL | 3mL | 3mL | 3mL |
| Antibiotic | 0 | 6µL of stock | 3µL of stock | 6µL of stock diluted 1/100 | 3µL of stock diluted 1/100 |
| Pre-culture | 300µL | 300µL | 300µL | 300µL | 300µL |
For each range, we test Pseudovibrio denitrificans but also E.Coli and E.Coli tranformed with a speicific resistance cassette (NM:The E.Coli strain for the control case is the same strain that is tranformed for the cassette efficiency test).
The initial OD at 600nm of each bacteria has been mesured to after be able to calculate the ΔOD at 600nm, during three days.
| / | Pseudo | Bl21 | DH5a | Top 10 | DH5a pyr+KanR | DH5a+CamR | Top 10+ErmR |
| OD init | 0.02 | 0.795 | 0.873 | 0.693 | 0.408 | 0.687 | 0.319 |
Plates for antibiotics's tests
For each tested antibiotic we made the same range of concentrations than in liquid.
We also made these range two time to have a replicate and we added a controle plate without antibiotic on which we showed all our tested bacteria. Plates will be checked during three days.
We made these same ranges of tested antibiotic's concentrations for three media:
- MB agar 1X
- MB agar 0.5X
- M9 agar 1X
Each plate is made like in the following table:
| / | 0 | 1:500 | 1:1000 | 1:50 000 | 1:100 000 |
| Medium+agar | 20mL | 20mL | 20mL | 20mL | 20mL |
| Antibiotic | 0 | 40µL of stock | 20µL of stock | 40µL of stock diluted 1/100 | 20µL of stock diluted 1/100 |
Aug 18
| Plates | LB Agar only | LB+Cam | LB+Kan | LB+Amp | LB+Erm | LB+Tet |
| E.coli | X | 0 | X | 0 | X | 0 |
There was a problem with the stock of Kanamycine so we made a new one at 25mg/mL.
For the erythrompycine, we learn that E. Coli is not really sensitive to this antibiotic and that it's better to use it with a dilution at 1:100.
Survivability tests - Results
| liquid cultures/Plates | MB | M9 |
| MB | ++++ | ++ |
| M9 | ++ | + |
Any liquid culture can be plated and will grow up. Of course, it works better on MB medium, wich is a rich medium than on M9.
Aug 17
Tests of antibiotics' stocks
Six plates of LB agar were made.
Five of them contained one of those antibiotics in the dilution 1:1000:
(The last one was the control of the growth of our bacteria without antibiotics) We sowed 10µL of a liquid LB culture of Bl21, and we let them incubate overnight at 37°C.
Survivability tests
Two plates of MB 1X and M9 1X were made and divised in two parts. Each plate was sowed with 5µL of a liquid MB 1X culture of Pseudovibrio denitrificans(dated 12th August) in a part and with 5µL of a liquid M9 1X culture Pseudovibrio denitrificans (dated 12th August) in the other part.
Pre-cultures
Cultures in MB 1X and M9 1X of Pseudovibrio were passed by a 1/10 dilution and let incubate overnight at 30°C. New cultures of different E.Coli were also started from glycerol or plate and let incubated overnight at 37°C.
From glycerol stocks:
From plates:
Bacteria tranformed with plasmid were cultivated with the corresponding antibiotic for the selection. For each medium we make a negative contrôle without bacteria. Aug 16
Tests for development of the electroporation's protocol
*Results of M9 1X plates - Day2:

Cold sorbitol seems to be the best way to wash our Pseudovibrio cells and avoid cells' death on M9 plates too.
*Results CFU:

Cold sorbitol is the best way to wash cells because it induce the bigest number of colonies on MB1X and bigger colonies on M91X.
Aug 13
Tests for development of the electroporation's protocol
*Results of M9 1X plates - Day1:

Results on M9 plates are not very readable.
*Results of MB 1X plates - Day1:

Cold sorbitol seems to be the best way to wash our Pseudovibrio cells and avoid cells' death on MB plates.
Aug 12
Tests for development of the electroporation's protocol
*Showed of MB Pseudovribrio electro-competent (which are stocked at -80°C) on MB 1X plates.
*Showed of M9 Pseudovribrio electro-competent (which are stocked at -80°C) on M9 1X plates.
Tests for development of the electroporation's protocol
Because of technical problems, we have to re-try with a culture of Pseudvirbrio in MB 1X.
Same protocol as 5th August.
Tests for development of the electroporation's protocol
> OD(600nm) in M9 = 0.15
> OD(600nm) in MB = 1.5
WORK IN ICE
Tests for development of the electroporation's protocol
*Pre-culture of Pseudovibrio denitrificans in 3mL of Marine broth 1X
*Pre-culture of Pseudovibrio denitrificans in 3mL of M9+CasAmino Acids 1X
Incubation overnight, 30°C.
Voltage tests

Curve of growth
We previously tried to af a curve of grotwh by mesuring the DO (600nm), but because of the opaqueness of the MB these data are useless.
We measured the growth of Pseudovibrio denitrificans on the TECAN in M9-CASA+3%NaCl and MB filtred (because of the opaqueness of the non filtred MB)


Unfortunately, cells didn't grow very well in the TECAN due to the little volume use and the ratio air/culture which is not optimal.
July 30
Construction of a new plasmid - Choice and amplification of the ORI
Purification of these PCR products.
Use “GenJEt” PCR Purification for it. The protocol is same that PCR purification except that the
binding buffer is add (1μL for 1mg) and boil at 55°C during 10 mn.
After the purification, a new PCR is realized with 3μL to purificated fragments.
Send to sequencing.
Construction of a new plasmid - Choice and amplification of the ORI
PCR with
Migration of the PCR products, we have two bands per well.

Figure: Result of ORI amplification in Pseudovibrio denitrificans (Pd), Pseudovribrio ascidiaceicola (Pa) and E.coli DH5a.
The negative control with E.coli DH5a confirms that there is no amplification. For Pseudovibrio strains, we obtain one band for Pseudovibrio denitrificans and two for Pseudovibrio ascidiaceicola.
July 23
Construction of a new plasmid - Choice and amplification of the ORI
We chose to amplify the genes RepA, RepB and RepC. We find these three genes with the genome browser on the genome of Pseudovibrio FOBEG1.
PCR with
Migration of the PCR products, we have no band the amplification didn't work.
July 22
Construction of a new plasmid - Verification of the promoter
A PCR was performed with primers 5 and 6 (see primers table in Protocols).
We purified those PCR product and sent them to be sequenced
Results :

July 21
Construction of a new plasmid - Choice and amplification of the promoter
We chose to amplify the tkt promoter of the transkelotase gene. We find this strong constitutive promoter with the genome browser on the genome of Pseudovibrio FOBEG1.
PCR with primers 5 and 6 (see primers table in Protocols), Q5 high fidelity enzyme and TM=55°C.
Transformation of Pseudovibrio with PSB1C3 (2) - Results
We have no colonies on the plates.
Transformation of Pseudovibrio with PSB1C3 (2)
We made electrocompetent Pseudovibrio and transform them by electroporation with
July 15
Transformation of Pseudovibrio with PSB1C3 - Results
We have no colonies on the plates.
Transformation of Pseudovibrio with PSB1C3
We made electrocompetent Pseudovibrio and transform them by electroporation with
July 12
Curve of growth
We try to measure a curve of grotwh by mesuring the DO (600nm) of a culture of Pseuovibrio denitrificans in MB 1X with a spectrometer

July 10
Protocols
Notebook - Protocols
Primers

RNA isolation (using TRI Reagent)
-Cleaning everything with RNA zap
-Switch on centrifuge for a fast cool at 4C
*Preparation of stabilisation buffer :
Add to 15 mL facon tube :
- 5ml Phenol
- 5ml 1M sodium acetate pH=5.5
Centrifuge 3 min, 4k rpm
Tranfer lower phase (contains phenol) to 50 mL falcon
Transfer 2.5mL to second falcon (devide into 2 tubes)
Add 45mL 100% ethanol to each tube
*Preparation of the samples
measurement of OD
if log phase, centrifugation of culture
Discard the supernatant
*Stabilization of samples
Add 1.25mL stabilization buffer to 10ml log phase culture
Put culture on ice
Vortex
Transfer into 15 ml falcon tube
Spin 4k rpm , 5 min
Discard supernatant
resuspend pellet in 1ml TRI Reagent
transfer to 2 ml eppie
incubate 5 min at RT
add 200µL chloroform
vortex 15 sec
incubate 15min at RT
Spin 12K rpm, 15min, 4C
transfer clear, aqueous phase (upper phase) to fresh tube
add 500µL isopropanol
vortex 5 sec
incubate 10' at RT
spin 14 000, 10 mn, 4C
carrefully pout off supernatant
add 1 mL 75% ethanol
spin 14000 rpm, 5min 4C
discard ethanol
air dry pellet 30 min
Put 93µL RNAse free water
Verification on an electrophoresis gel (caution: preparation of electrophoresis gel with TBE or TAE RNAse Free agarose 1%)
Measuring quantity of RNA with nanodrop
Turbo DNAse protocol of RNA
Dilute sample to 200ng/µL in 100µL (20µg total)
Add 10µL 10X DNAse buffer
Add 2µL turbo Dnase (2U/µL)
Incubate at 37C, 30min
*Phenol-Chloroform extraction
Ajust the volume of this sample at 200µL
Add 1 volume of phonol:chloroform:isomyl alcohol
Vortex
spin 14K , 5min, 4C
Transfer upper , aqueous phase to fresh tube
Verification on an electrophoresis gel (if DNA IS ALWAYS present, do again the step of Turbo DNAse treatment)
*Protocol for ethanol/acetate precipitation of RNA
if small volume, bring to 180µL with RNAse-free water
add 0.1 volume 5M ammonium acetate or 3M sodium acetate
(optional) add 2µL of 5µg/µL glycogen if RNA is < 200µg/µL
add 2.5-3 volumes 100% ethanol
put at -20C o/n or -80C for 30min
13K, 30min, 4C
carrefully discard supernatant
add 1ml ice cold 70% ethanol
13K, 10min, 4C
discard ethanol air dry 10min
Resuspend RNA in RNAse-free water(max 18µL)
Verification on an electrophoresis gel
*Nanodrop
*RNAship
DNA extraction
(link here)
Transformation of Pseudovibrio
- Place electroporation tanks in ice for 10min
- Take samples of plasmids (25ng-50ng/µL) /!\ Keep them in ice /!\
- Take sample of competent cells /!\ Keep them in ice /!\
- Place 1µL of plasmid in the sample of competent cells
- Transfer the full volume obtained in the electroporation tank
- Place in the electroporator and pulse at 2000V NB: The optimal pulse length is between 5 and 6ms.
- Add 1mL of MB 1X in the 30 seconds following the transformation
- Incubate between 2h and 3h at 30°C with shaking
- Centrifuge to concentrate all cells in the pellet
- Discard the supernatant
- Sowed the pellet on selective plates of MB 1X
Electro-competent Pseudovibrio in Marine broth
* Refrigerated centrifuge (4°C)
* Marine Broth 1X
*Cold glycerol 10% (4°C)
Experimental procedure
-Make a pre-culture of Pseudovibrio in 3mL of MB 1X and let it incubate overnight at 30°C with shaking.
-Relaunch the Pseudovibrio culture in 100mL of MB 1X.
-Let it grow in a 30°C shaking incubator until DO (600nm) = 1.2
-Divide the culture into two 50mL Falcon tubes.
-Wash cells with cold glycerol 10% five times
(Centrifuge at 4000g, 4°C for 10min and re-suspend):
* 50 mL (x 1)
* 25 mL (x 1)
* 5 mL (x 3)
-Re-suspend a last time washed cells in 500µL of glycerol 10%.
-Aliquot 50µL of competent cells and stock them at -80°C for maximum three weeks.
Electro-competent Pseudovibrio in M9-CASA +3%NaCl
* Refrigerated centrifuge (4°C)
* M9-CASA+3%NaCl 1X
*Cold glycerol 10% (4°C)
*Cold sorbitol 2% (4°C)
Experimental procedure
-Make a pre-culture of Pseudovibrio in 3mL of M9-CASA+3%NaCl 1X and let it incubate overnight at 30°C with shaking.
-Relaunch the Pseudovibrio culture in 100mL of M9-CASA+3%NaCl 1X.
-Let it grow in a 30°C shaking incubator until DO (600nm) = 1.2
-Divide the culture into two 50mL Falcon tubes.
-Wash cells with cold glycerol or cold sorbitol three times
(Centrifuge at 4000g, 4°C for 10min and re-suspend):
* 50 mL (x 1)
* 25 mL (x 1)
* 5 mL (x 1)
-Re-suspend a last time washed cells in 500µL of glycerol 10%.
-Aliquot 50µL of competent cells and stock them at -80°C for maximum three weeks.
Marine broth
Composition of the medium:

After having put the wished volume of water, put 40.2g/L of the medium powder.
Dissolve sediments by warming the mixture.
Boil during one entiere minute
Autoclave the medium during 15min at 250°F.
M9 - CASA +3%NaCl
Composition of the medium:

After add correspondant quantity of the different compounds in the wished volume of water, filtrate the entiere volume obtained.
NB: This medium can't be autoclaved contain glucose and amino acids.
Preparation of antibiotic stocks
| Antibiotic | Stock concentration | Protocol |
| Kanamycin | 25 mg/mL | Weight 0.56g of Kanamycin powder, solubilization into 20mL of miliQ water. Vortex 5min and filtration with a 200nm filter. Stock : -20°C (aliquots of 1mL) |
| Streptomycin | 100 mg/mL | Weight 1g of Streptomycin powder, solubilization into 20mL of miliQ water. Vortex 5min and filtration with a 200nm filter. Stock : -20°C (aliquots of 1mL) |
| Amplicilin | 100 mg/mL | Weight 1g of Amplicilin powder, solubilization into 20mL of miliQ water. Vortex 5min and filtration with a 200nm filter. Stock : -20°C (aliquots of 1mL) |
| Tetracyclin | 15 mg/mL | Weight 0.6g of Tetracyclin powder, solubilization into 20mL of ethanol 50%. Vortex 5min and filtration with a 200nm filter. Stock : -20°C and hiden from light(aliquots of 1mL) |
| Chloramphenicol | 34 mg/mL | Weight 0.34g of Chloramphenicol powder, solubilization into 10mL of ethanol 100%. Vortex 5min and filtration with a 200nm filter. Stock : -20°C and hiden from light(aliquots of 1mL) |