"
"
Team:SUSTC-Shenzhen/Project
From 2014.igem.org
(→Application in other retroviral diseases) |
|||
| Line 4: | Line 4: | ||
{{:Team:SUSTC-Shenzhen/templates/page-header| | {{:Team:SUSTC-Shenzhen/templates/page-header| | ||
| - | title= | + | title=Cell Line Construction| |
| - | subtitle= | + | subtitle= Brief summary of cell line construction!}} |
| + | |||
| + | {{SUSTC-Math-Enabled}} | ||
{{SUSTC-Shenzhen/main-content-begin}} | {{SUSTC-Shenzhen/main-content-begin}} | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | + | ==Introduction== | |
| - | + | The fancy idea to equip human cells with an intracellular self-protecting immune system against lentivirus especially HIV is about to realize by introducing Cas9 protein into the genome. | |
| + | However, does Cas9 protein have sufficient efficiency to disrupt targeted virus DNA sequence and disable it to free patient from lentivirus caused disease. Does the side effect acceptable. These still remain unknown questions, need more experiments to determine. | ||
| + | Our strategy to cure and prevent AIDS is to integrate Cas9 gene into hematopoietic stem cells. When HIV infects human, plasmid encoding appropriate gRNA sequence targeting HIV sequence will be introduced into the transformed stem cells, then HIV genome will be cut off by Cas9 protein. | ||
| - | |||
| - | == | + | ==Experiment Design== |
| - | + | ||
| - | + | ||
| - | + | Will it works? | |
| - | + | To verify we design 3 steps of experiments. | |
| - | + | ||
| - | == | + | ===The first step:=== |
| - | + | We use HeLa cells and EGFP to simplify the experiment because using hematopoietic stem cells and HIV is impractical for us. We will test the idea in HeLa cells which is a common used immortal human cell line (wikipedia) and the EGFP gene will be a substitute of HIV gene firstly. Both Cas9 gene and EGFP gene are stably transfected into HeLa cells, which mimics that the hematopoietic stem cell equipped with Cas9 are infected by HIV. Then the plasmid encoding gRNA targeting EGFP gene is introduced into the cells to measure the efficiency of the Cas9 protein to cut off the targeted sequence with the guide of gRNA. | |
| - | + | ||
| - | + | ||
| - | == | + | ===The second step:=== |
| - | + | Whether the targeted sequences of HIV screened out by our team can be efficiently disrupted and safety of our target sequence will be tested. To make the result easier to detect and measure quantitatively, the virus sequences will be inserted into EGFP gene by locating the sequence before or on the both ends of the EGFP gene. Then the plasmid encoding gRNA to target virus sequence will be introduced into the cells to see whether the virus sequence can be disrupted or eradicated successfully. | |
| - | + | ===The third step:=== | |
| - | + | The third part is A-B toxin delivery, to respond to the high mutation rate of HIV. In other words, different gRNAs will be delivered into the helper T cells of the patient whose hematopoietic stem cells have been equipped with Cas9 gene. A safety In vitro delivery of DNA into targeted cells specifically is needed. In our project, two fusion proteins TEG and GD5 which contain a domain mimics bacteria A-B toxin are used for DNA delivery. | |
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | + | ==The construction of HeLa cell line stably transfected with EGFP gene and Cas9 gene.== | |
| - | + | ||
| - | <center>{{SUSTC-Image|wiki/images/ | + | To obtain a cell line with EGFP gene and Cas9 gene integrated in, we used PiggyBac transposon system to stably transfect. Then expand mono-clones and population expressing resistance into cell line. |
| - | + | <center>{{SUSTC-Image|wiki/images/0/06/SUSTC-Shenzhen-Project-PiggyBac-transposon.png}}</center> | |
| + | Figure 1. Mechanism of PiggyBac transposon system | ||
| + | It is estimated that only part of the transfected cells will be integrated with Cas9 gene and EGFP gene. Resistance screening we have used to get a pure cell pool of transfected cells. Plasmid Cas9-TetOn expressing Cas9 under the control of Tet-ON 3G using Piggybac transposon system encodes puromycin resistance, and plasmid PB-EGFP to stably express EGFP in host cells using Piggybac transposon system encodes blasticidin resistance.<html><a href="https://2014.igem.org/Team:SUSTC-Shenzhen/Notebook/HeLaCell/PB5-TetOn-Cas9-PB3-plasmid-construction">Detail about plasmid construction</a></html> | ||
| - | < | + | <center>{{SUSTC-Image|wiki/images/1/17/SUSTC-Shenzhen-Project-PB-TetON-Cas9.jpg}}</center> |
| - | + | Figure 2. PB-TetON-Cas9 plasmid, PB5-PB3 are transposon, Puro is resistance gene. | |
| - | + | ||
| - | + | <center>{{SUSTC-Image|wiki/images/7/73/SUSTC-Shenzhen-Project-PB-EGFP.png}}</center> | |
| - | + | Figure 3. PB-EGFP plasmid, PB5-PB3 are transposon, Bla is resistance gene. | |
| - | + | ||
| - | <center>{{SUSTC-Image|wiki/images/ | + | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | To determine the minimum usage level of puromycin and blasticidin in culture medium to inhibit the growth of normal HeLa cell, we culture cells in medium with gradient concentration in different wells of cell culture plate. We found that the minimum efficient concentration of bla(blasticidin) is 2.8μg/mL <html><a href="https://2014.igem.org/Team:SUSTC-Shenzhen/Notebook/HeLaCell/Determining-the-appropriate-concentration-of-blasticidin-for-cell-selection" style="color:999">(More Details)</a></html>and puro(puromycin) is 2.0μg/mL.<html><a href="https://2014.igem.org/Team:SUSTC-Shenzhen/Notebook/HeLaCell/Determining-the-appropriate-concentration-of-puromycin-for-cell-selection" style="color:999">(More Details)</a></html> | |
| - | + | ||
| - | + | After about one month selection, the percentage of HeLa cells expressing EGFP was about 95% (Figure 4 & 5). | |
| - | + | ||
| - | + | <center>{{SUSTC-Image|wiki/images/b/b7/SUSTC-Shenzhen-G1-before-freeze.png|G1 pool before freezing}}</center> | |
| - | + | Figure 4. Pool cells transfected with EGFP gene before freezing. | |
| - | <center>{{SUSTC-Image|wiki/images/ | + | <center>{{SUSTC-Image|wiki/images/c/ca/SUSTC-Shenzhen-G3-before-freeze.png|G3 pool before freezing}}</center> |
| - | + | Figure 5. Pool cells transfected with EGFP gene and Cas9 gene before freezing | |
| - | + | Monoclonal cells are supposed to have the same insertion sites and copy number which make the system start with fewer variables. Moreover, monoclonal cells with low copy number of EGFP gene is preferred and constant copy number of Cas9 gene. Cells with high copies of EGFP gene may conceal the damage of part of them caused by Cas9 and therefore underestimate the efficiency of Cas9 protein and gRNA. | |
| - | + | ||
| - | + | So we have also cultured some monoclonal cells whose fluorescence intensity appeared to be lower. Comparing with the pool of the cells stably transfected with EGFP gene and Cas9 gene, the monoclonal cells has a smaller EGFP intensity range. Then the several monoclonal cells are transfected with plasmid encoding the gRNA for EGFP. On the whole, the percentage of the cells with EGFP signal in all clones transfected with plasmid encoding gRNA decline dramatically, decline from 91%- 99% to 50%-79%(Table 1, Figure6).<html> | |
| - | + | <a href="https://2014.igem.org/Team:SUSTC-Shenzhen/Notebook/HeLaCell/The_quality_test_of_the_clones_of_G3%26G4" style="color:999">(More Details)</a></html> | |
| - | < | + | <center>{{SUSTC-Image|wiki/images/1/1c/SUSTC-Shenzehn-clone_4-2_and_G3_cytometry_change.png|G3 4-2 flow cytometry}}</center> |
| - | + | Figure 6. Flow cytometry reading pool cells(G3) and monoclones(4-2). | |
| - | + | ||
| - | = | + | <html><div class="table-responsive"></html> |
| - | + | {| class="table" | |
| + | |+ style="font-size:18px; font-weight:bold"|Summary of the data of flow cytometry | ||
| + | ! Clones | ||
| + | ! The change of the percentage EGFP signal | ||
| + | ! The change of the mean of the intensity of the EGFP signal of the positive cells | ||
| + | |- | ||
| + | | A1 | ||
| + | | 91% to 79% | ||
| + | | 34 to 40 | ||
| + | |- | ||
| + | | A2 | ||
| + | | 91% to 79% | ||
| + | | 35 to 18 | ||
| + | |- | ||
| + | | B5 | ||
| + | | 97% to 57% | ||
| + | | 33 to 19 | ||
| + | |- | ||
| + | | C5 | ||
| + | | 99% to 67% | ||
| + | | 34 to 17 | ||
| + | |- | ||
| + | | 4-2 | ||
| + | | 98% to 64% | ||
| + | | 28 to 20 | ||
| + | |- | ||
| + | | G3 pool | ||
| + | | 98% to 67% | ||
| + | | 53 to 39 | ||
| + | |} | ||
| + | <html></div></html> | ||
| + | Table 1. The summary of the data of flow cytometry, reading monoclones integrated with Cas9 and EGFP gene. | ||
| - | + | ==The construction of HeLa cell line stably transfected with modified EGFP== | |
| - | + | To test Cas9 cleave viral DNA, and to screen the result of cleavage, we designed 3 plasmid. Insert target sequence is near the original EGFP sequence between poly A and promoter, so it will be transcripted into mRNA. The target sequence is in the reading frame in synthesis protein. The consequence of lost base pair leads to frame shift mutation, thus disfunction the protein. | |
| - | + | {{SUSTC-Image|wiki/images/6/60/SUSTC-Shenzhen-PB-EGFP-HIV1.png|}} | |
| + | Figure 7. Plasmid of EGFP+HIV1 viral sequence | ||
| + | {{SUSTC-Image|wiki/images/5/52/SUSTC-Shenzhen-PB-EGFP-HBV12.png|}} | ||
| + | Figure 8. Plasmid of EGFP+HBV1.2 viral sequence | ||
| + | {{SUSTC-Image|wiki/images/2/2a/SUSTC-Shenzhen-PB-EGFP-HBV2.png|}} | ||
| + | Figure 9. Plasmid of EGFP+HBV2 viral sequence | ||
| - | + | After Cas9 cleave the target sequence, it is easy to be repair by error-prone Non-Homologous-Recombination(NHR)(F Ann Ran et al., 2013), which is easy to leave a scar and cause protein frame-shift mutation.(Prashant et al., 2013) Targeting the viral sequence and induce EGFP mutation mimics Cas9 targeting viral genome in human cells, and silence the expression of crucial protein of virus. When Cas9 cut the viral DNA the double cleave of DNA would cause NHR, so that the DNA sequence is easily mutated. | |
| - | + | We used the way of construct cell line expressing EGFP to construct cell line with EGFP+target sequence. Before wiki due day, we still do not finish the selection of cell-line. But we still introduced plasmid encoding gRNA targeting viral sequence. <html><a href="https://2014.igem.org/Team:SUSTC-Shenzhen/Notebook/HeLaCell/Stably_transfect_cell_with_target-sequence_EGFP">For details</a></html> | |
| - | + | We have read the result Oct.16th 11days after transfection by flow cytometry. | |
| - | + | {{SUSTC-Image|wiki/images/c/c7/SUSTC-Shenzhen-target-sequence-cell-line-construct-figure10.png|Flow Cytometry result}} | |
| + | Figure 10. | ||
| - | + | <html><div class="table-responsive"></html> | |
| + | {| class="table" | ||
| + | |+ style="font-size:18px; font-weight:bold"|Summary of the data of flow cytometry | ||
| + | ! Clones | ||
| + | ! Ratio of positive | ||
| + | |- | ||
| + | | HIV1 | ||
| + | | 60% | ||
| + | |- | ||
| + | | HBV1.2 | ||
| + | | 1.11% | ||
| + | |- | ||
| + | | HBV2 | ||
| + | | 63% | ||
| + | |} | ||
| + | <html></div></html> | ||
| + | table 2. Summary of flow cytometry result. The fraction of postive is very low in group HBV1.2, because the NLS is disfunctioned by two inserted viral sequence. | ||
| - | |||
| - | + | ==The A-B toxin transfection== | |
| + | The Cas9 and gRNA system has been proved to disrupt the targeted EGFP gene efficiently. It comes out to our final part: to deliver plasmids encoding gRNA into cells. In this part two fusion proteins TEG and GD5 will be used to do transfection. TEG is a fusion protein designed for intracellular delivery of DNA by binding to the EGF receptor on human cells. It incorporates GAL4 which can bind to plasmids with UAS sequence, TGF-α which is a ligand of human EGF receptor and sequences encoding the translocation domain of Pseudomonas exotoxin A. GD-5 is another fusion protein similar to TEG and it mediates the DNA transfection by binding to ErbB2 receptor on human cells. | ||
| - | + | The proteins are expressed in E.coli, purified and renatured. It is a challenge work. The two proteins were purified and renatured several times and so was the cells transfection with the two fusion proteins. We were very excited when the first batch of TEG protein renatured was obtained and the transfection is carried out soon. The procedure of the transfection is designed mainly according to the two original paper of the two fusion proteins. 0.5 nM DNA, 25nM and 50-160 nM poly-lysine HBr are the final concentration suggested. DNA and protein solution are mixed and poly-lysine HBr is slowly added to condensate DNA-protein complex. The UAS plasmids encoding both gRNA and mCherry were transfected into the cells using TEG protein. The protocol is basically consist with the method suggested. However, we failed to observe the mcherry signal under the fluorescence microscope. Then we used new batches of proteins obtained to do transfection. And the transfection procedure was improved by increasing the amount of DNA and protein. What’s more, considering the EGF in the FBS may antagonize the TEG protein and GD5 protein, this time part of cells transfected are cultured in DMEM without FBS during transfection. And different cell density is also tried. To increase the sensitivity of mCherry signal detection, the cells transfected were also read by a flow cytometry in addition to the observation under fluorescence microscope. Unfortunately, we have not found valid mCherry signal in the transfected cells (figures). | |
| - | + | ||
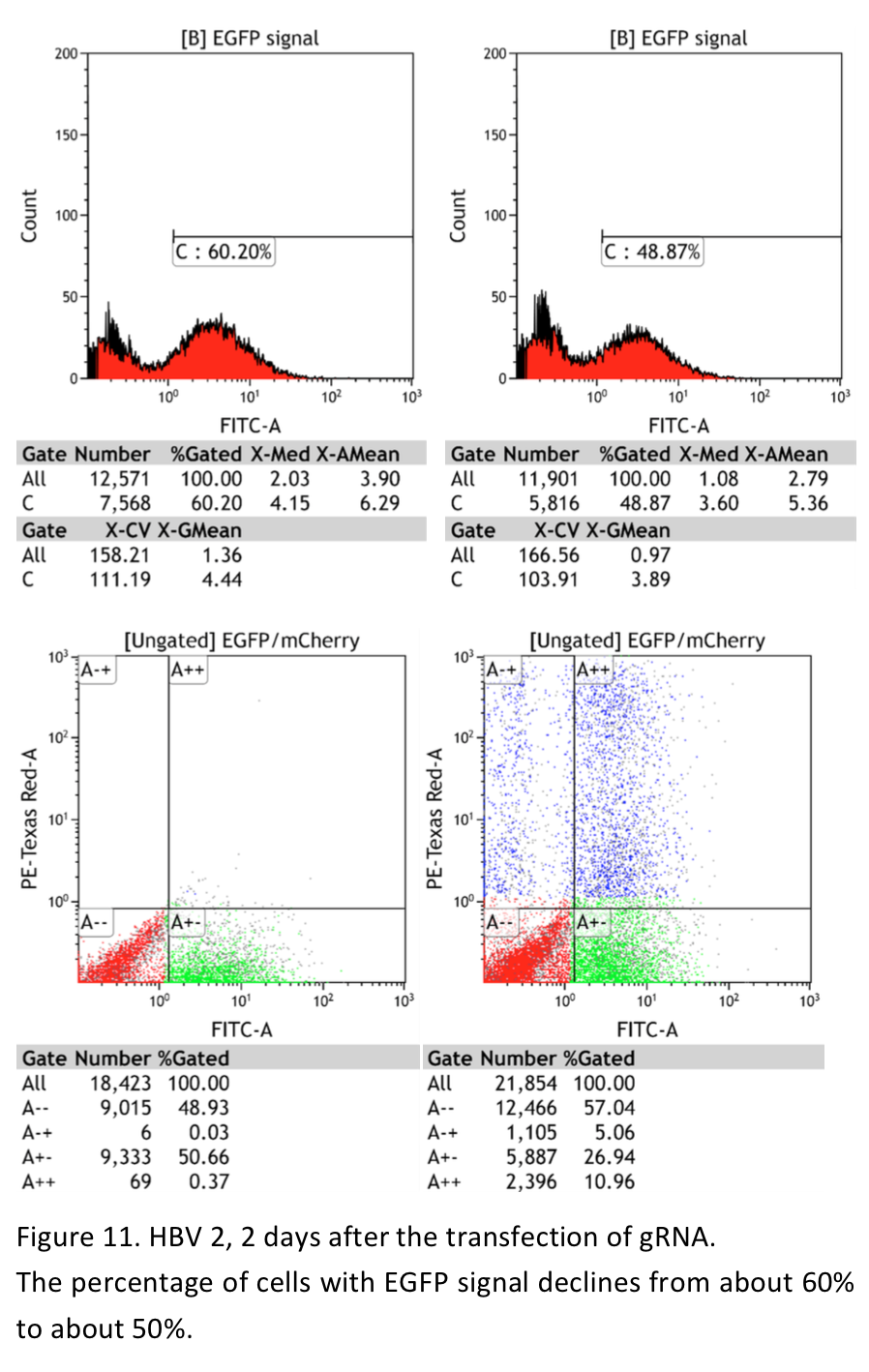
| - | + | {{SUSTC-Image|wiki/images/0/00/SUSTC-Shenzhen-target-sequence-cell-line-construct-figure11.png|Figure 11.}} | |
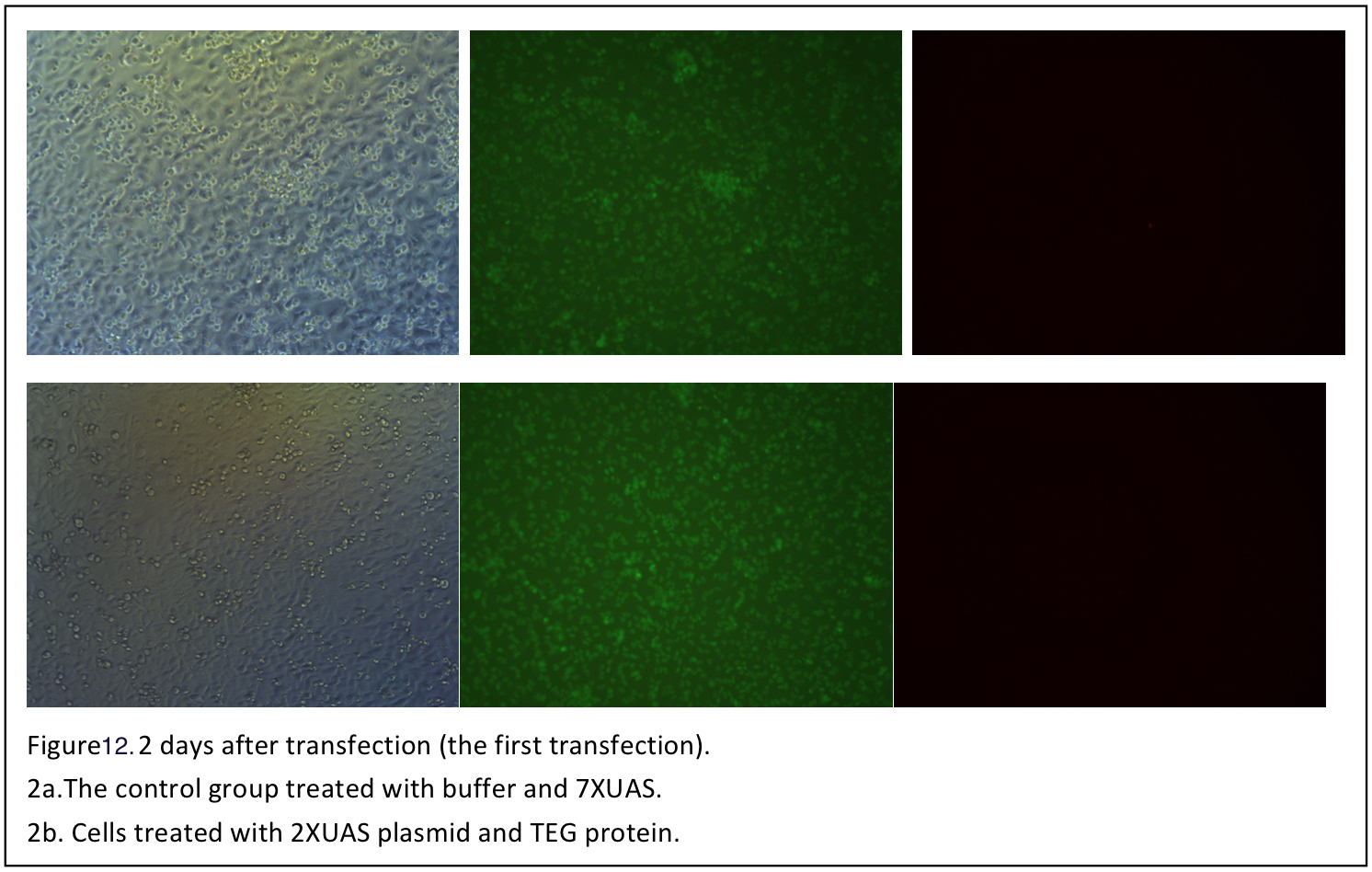
| + | {{SUSTC-Image|wiki/images/8/8f/SUSTC-Shenzhen-A-B-Toxin-figure12.jpg|Figure 12.}} | ||
{{SUSTC-Shenzhen/main-content-end}} | {{SUSTC-Shenzhen/main-content-end}} | ||
{{SUSTC-Shenzhen/wiki-footer}} | {{SUSTC-Shenzhen/wiki-footer}} | ||
{{SUSTC-Shenzhen/themeJs}} | {{SUSTC-Shenzhen/themeJs}} | ||
Revision as of 03:49, 18 October 2014
Cell Line Construction
Brief summary of cell line construction!
Contents |
Introduction
The fancy idea to equip human cells with an intracellular self-protecting immune system against lentivirus especially HIV is about to realize by introducing Cas9 protein into the genome. However, does Cas9 protein have sufficient efficiency to disrupt targeted virus DNA sequence and disable it to free patient from lentivirus caused disease. Does the side effect acceptable. These still remain unknown questions, need more experiments to determine. Our strategy to cure and prevent AIDS is to integrate Cas9 gene into hematopoietic stem cells. When HIV infects human, plasmid encoding appropriate gRNA sequence targeting HIV sequence will be introduced into the transformed stem cells, then HIV genome will be cut off by Cas9 protein.
Experiment Design
Will it works? To verify we design 3 steps of experiments.
The first step:
We use HeLa cells and EGFP to simplify the experiment because using hematopoietic stem cells and HIV is impractical for us. We will test the idea in HeLa cells which is a common used immortal human cell line (wikipedia) and the EGFP gene will be a substitute of HIV gene firstly. Both Cas9 gene and EGFP gene are stably transfected into HeLa cells, which mimics that the hematopoietic stem cell equipped with Cas9 are infected by HIV. Then the plasmid encoding gRNA targeting EGFP gene is introduced into the cells to measure the efficiency of the Cas9 protein to cut off the targeted sequence with the guide of gRNA.
The second step:
Whether the targeted sequences of HIV screened out by our team can be efficiently disrupted and safety of our target sequence will be tested. To make the result easier to detect and measure quantitatively, the virus sequences will be inserted into EGFP gene by locating the sequence before or on the both ends of the EGFP gene. Then the plasmid encoding gRNA to target virus sequence will be introduced into the cells to see whether the virus sequence can be disrupted or eradicated successfully.
The third step:
The third part is A-B toxin delivery, to respond to the high mutation rate of HIV. In other words, different gRNAs will be delivered into the helper T cells of the patient whose hematopoietic stem cells have been equipped with Cas9 gene. A safety In vitro delivery of DNA into targeted cells specifically is needed. In our project, two fusion proteins TEG and GD5 which contain a domain mimics bacteria A-B toxin are used for DNA delivery.
The construction of HeLa cell line stably transfected with EGFP gene and Cas9 gene.
To obtain a cell line with EGFP gene and Cas9 gene integrated in, we used PiggyBac transposon system to stably transfect. Then expand mono-clones and population expressing resistance into cell line.
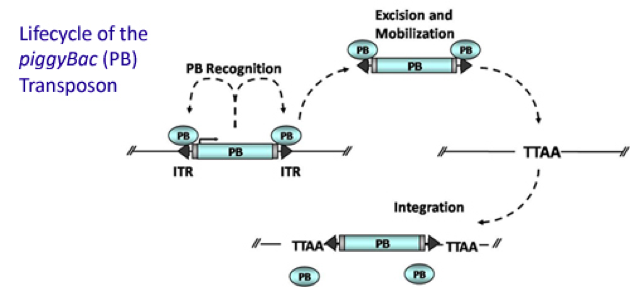
Figure 1. Mechanism of PiggyBac transposon system
It is estimated that only part of the transfected cells will be integrated with Cas9 gene and EGFP gene. Resistance screening we have used to get a pure cell pool of transfected cells. Plasmid Cas9-TetOn expressing Cas9 under the control of Tet-ON 3G using Piggybac transposon system encodes puromycin resistance, and plasmid PB-EGFP to stably express EGFP in host cells using Piggybac transposon system encodes blasticidin resistance.Detail about plasmid construction
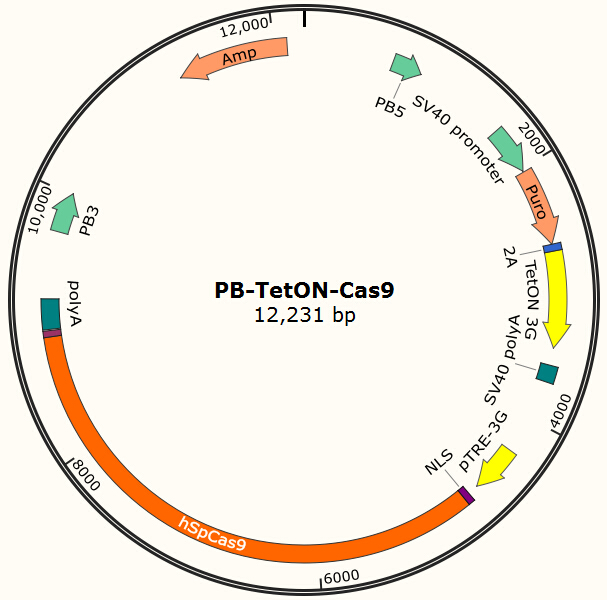
Figure 2. PB-TetON-Cas9 plasmid, PB5-PB3 are transposon, Puro is resistance gene.
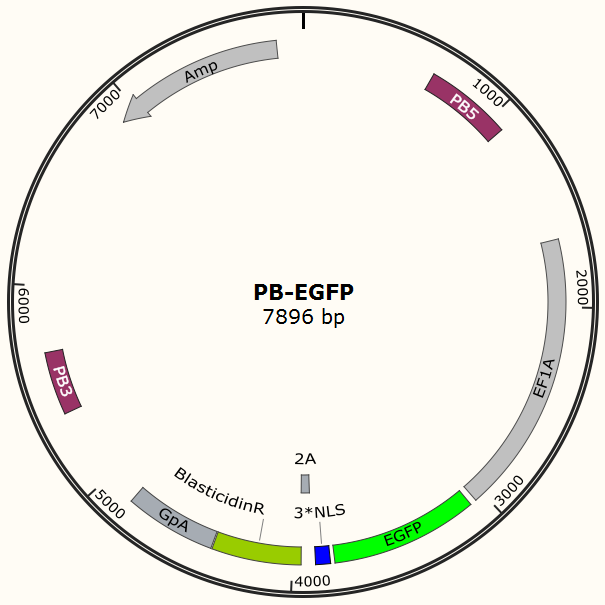
Figure 3. PB-EGFP plasmid, PB5-PB3 are transposon, Bla is resistance gene.
To determine the minimum usage level of puromycin and blasticidin in culture medium to inhibit the growth of normal HeLa cell, we culture cells in medium with gradient concentration in different wells of cell culture plate. We found that the minimum efficient concentration of bla(blasticidin) is 2.8μg/mL (More Details)and puro(puromycin) is 2.0μg/mL.(More Details)
After about one month selection, the percentage of HeLa cells expressing EGFP was about 95% (Figure 4 & 5).
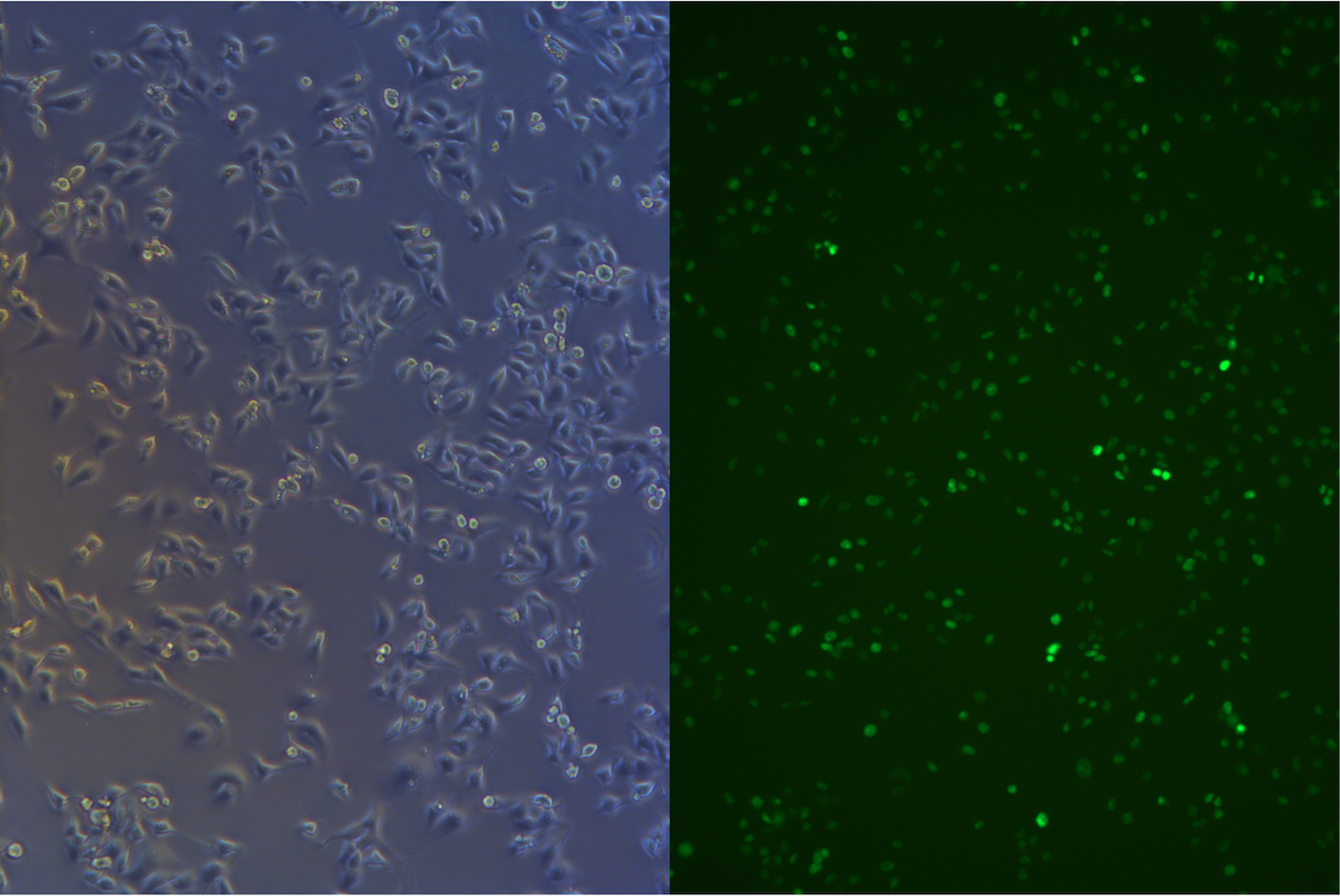
Figure 4. Pool cells transfected with EGFP gene before freezing.
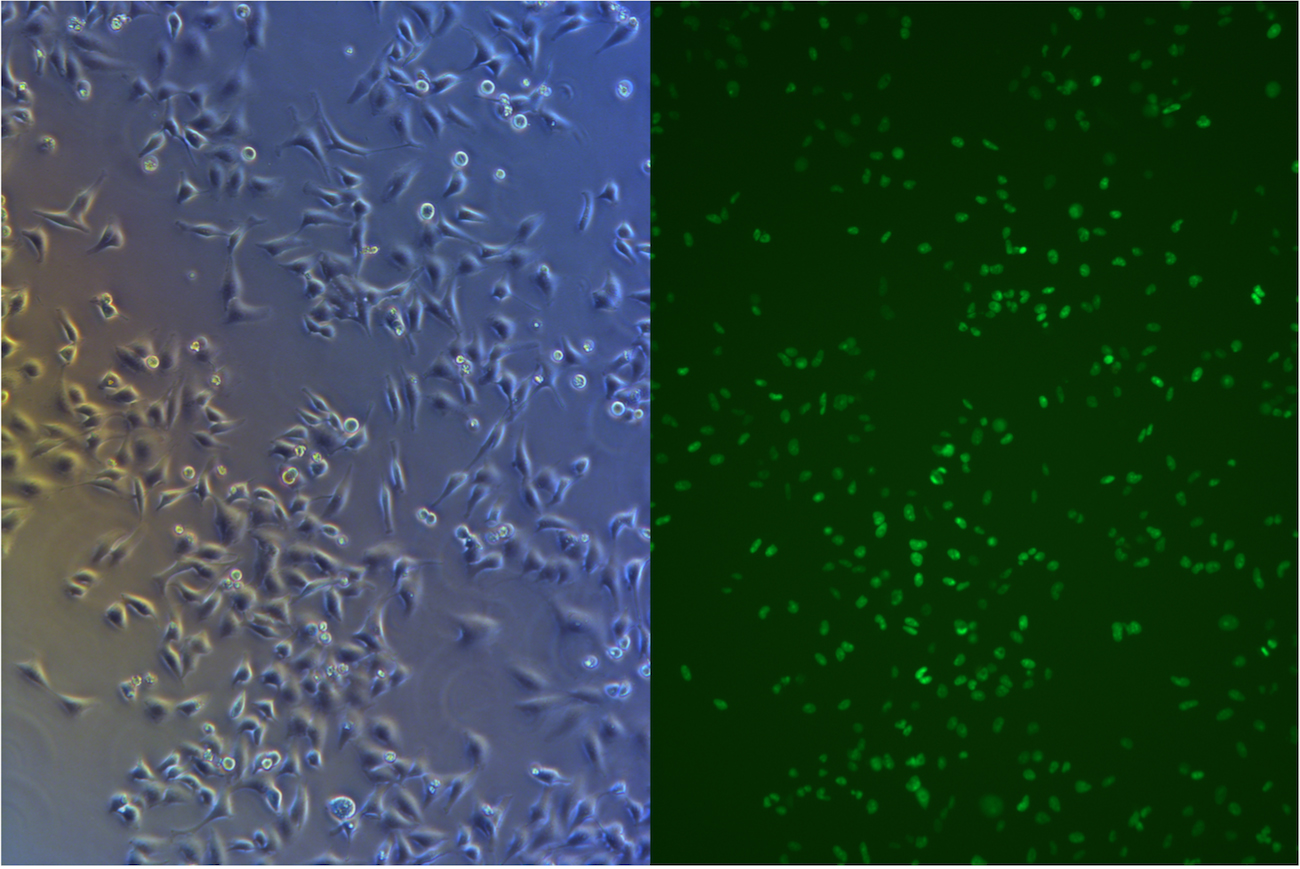
Figure 5. Pool cells transfected with EGFP gene and Cas9 gene before freezing
Monoclonal cells are supposed to have the same insertion sites and copy number which make the system start with fewer variables. Moreover, monoclonal cells with low copy number of EGFP gene is preferred and constant copy number of Cas9 gene. Cells with high copies of EGFP gene may conceal the damage of part of them caused by Cas9 and therefore underestimate the efficiency of Cas9 protein and gRNA.
So we have also cultured some monoclonal cells whose fluorescence intensity appeared to be lower. Comparing with the pool of the cells stably transfected with EGFP gene and Cas9 gene, the monoclonal cells has a smaller EGFP intensity range. Then the several monoclonal cells are transfected with plasmid encoding the gRNA for EGFP. On the whole, the percentage of the cells with EGFP signal in all clones transfected with plasmid encoding gRNA decline dramatically, decline from 91%- 99% to 50%-79%(Table 1, Figure6). (More Details)
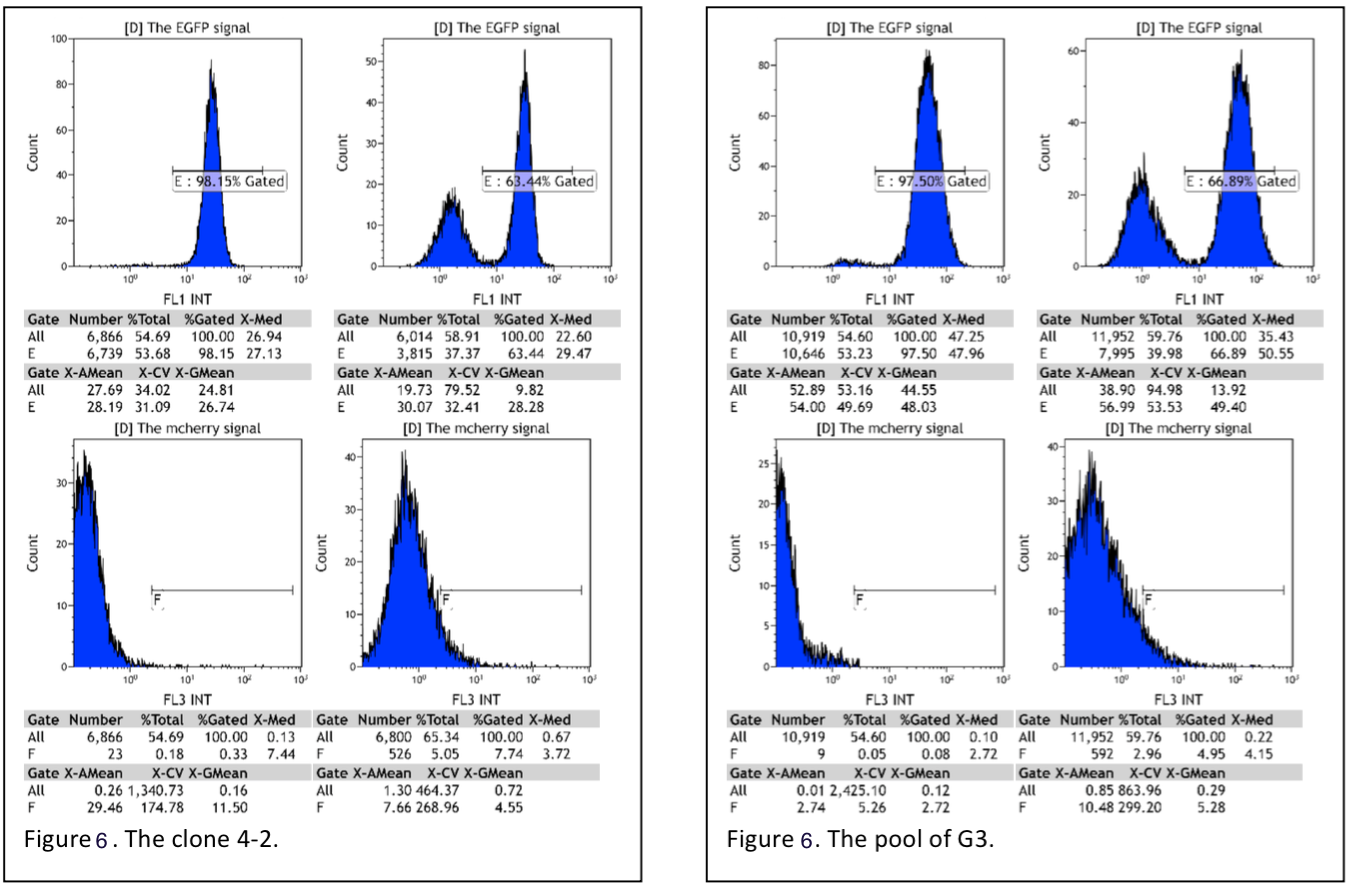
Figure 6. Flow cytometry reading pool cells(G3) and monoclones(4-2).
| Clones | The change of the percentage EGFP signal | The change of the mean of the intensity of the EGFP signal of the positive cells |
|---|---|---|
| A1 | 91% to 79% | 34 to 40 |
| A2 | 91% to 79% | 35 to 18 |
| B5 | 97% to 57% | 33 to 19 |
| C5 | 99% to 67% | 34 to 17 |
| 4-2 | 98% to 64% | 28 to 20 |
| G3 pool | 98% to 67% | 53 to 39 |
Table 1. The summary of the data of flow cytometry, reading monoclones integrated with Cas9 and EGFP gene.
The construction of HeLa cell line stably transfected with modified EGFP
To test Cas9 cleave viral DNA, and to screen the result of cleavage, we designed 3 plasmid. Insert target sequence is near the original EGFP sequence between poly A and promoter, so it will be transcripted into mRNA. The target sequence is in the reading frame in synthesis protein. The consequence of lost base pair leads to frame shift mutation, thus disfunction the protein.

Figure 7. Plasmid of EGFP+HIV1 viral sequence

Figure 8. Plasmid of EGFP+HBV1.2 viral sequence

Figure 9. Plasmid of EGFP+HBV2 viral sequence
After Cas9 cleave the target sequence, it is easy to be repair by error-prone Non-Homologous-Recombination(NHR)(F Ann Ran et al., 2013), which is easy to leave a scar and cause protein frame-shift mutation.(Prashant et al., 2013) Targeting the viral sequence and induce EGFP mutation mimics Cas9 targeting viral genome in human cells, and silence the expression of crucial protein of virus. When Cas9 cut the viral DNA the double cleave of DNA would cause NHR, so that the DNA sequence is easily mutated.
We used the way of construct cell line expressing EGFP to construct cell line with EGFP+target sequence. Before wiki due day, we still do not finish the selection of cell-line. But we still introduced plasmid encoding gRNA targeting viral sequence. For details
We have read the result Oct.16th 11days after transfection by flow cytometry.
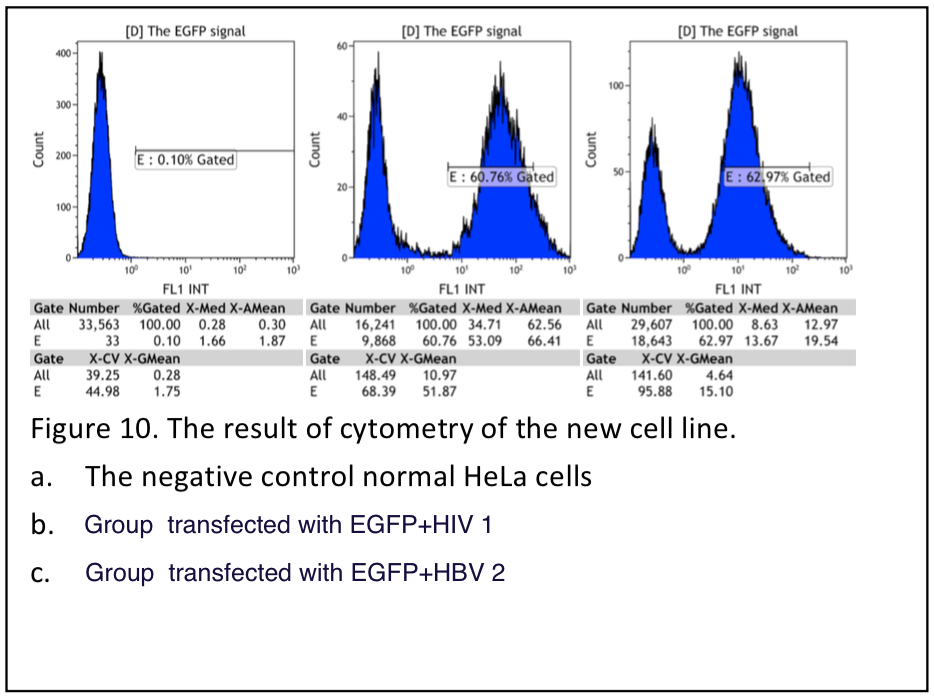
Figure 10.
| Clones | Ratio of positive |
|---|---|
| HIV1 | 60% |
| HBV1.2 | 1.11% |
| HBV2 | 63% |
table 2. Summary of flow cytometry result. The fraction of postive is very low in group HBV1.2, because the NLS is disfunctioned by two inserted viral sequence.
The A-B toxin transfection
The Cas9 and gRNA system has been proved to disrupt the targeted EGFP gene efficiently. It comes out to our final part: to deliver plasmids encoding gRNA into cells. In this part two fusion proteins TEG and GD5 will be used to do transfection. TEG is a fusion protein designed for intracellular delivery of DNA by binding to the EGF receptor on human cells. It incorporates GAL4 which can bind to plasmids with UAS sequence, TGF-α which is a ligand of human EGF receptor and sequences encoding the translocation domain of Pseudomonas exotoxin A. GD-5 is another fusion protein similar to TEG and it mediates the DNA transfection by binding to ErbB2 receptor on human cells.
The proteins are expressed in E.coli, purified and renatured. It is a challenge work. The two proteins were purified and renatured several times and so was the cells transfection with the two fusion proteins. We were very excited when the first batch of TEG protein renatured was obtained and the transfection is carried out soon. The procedure of the transfection is designed mainly according to the two original paper of the two fusion proteins. 0.5 nM DNA, 25nM and 50-160 nM poly-lysine HBr are the final concentration suggested. DNA and protein solution are mixed and poly-lysine HBr is slowly added to condensate DNA-protein complex. The UAS plasmids encoding both gRNA and mCherry were transfected into the cells using TEG protein. The protocol is basically consist with the method suggested. However, we failed to observe the mcherry signal under the fluorescence microscope. Then we used new batches of proteins obtained to do transfection. And the transfection procedure was improved by increasing the amount of DNA and protein. What’s more, considering the EGF in the FBS may antagonize the TEG protein and GD5 protein, this time part of cells transfected are cultured in DMEM without FBS during transfection. And different cell density is also tried. To increase the sensitivity of mCherry signal detection, the cells transfected were also read by a flow cytometry in addition to the observation under fluorescence microscope. Unfortunately, we have not found valid mCherry signal in the transfected cells (figures).