 "
"
Team:SUSTC-Shenzhen/Project
From 2014.igem.org
(→Constructing EGFP-Cas9 expressing cell line) |
|||
| (16 intermediate revisions not shown) | |||
| Line 24: | Line 24: | ||
=Background= | =Background= | ||
==HIV (Human immunodeficiency virus)== | ==HIV (Human immunodeficiency virus)== | ||
| - | + | '''HIV''' is the trigger of the lethal disease '''AIDS'''. It replicates in and kills '''T cells''', weakens human immune system and allows life-threatening opportunistic infection and cancer to thrive. HIV infects vital cells in the human immune system such as helper T cells (especially CD4+ T cells), macrophages, and dendritic cells. As a retrovirus, HIV reverse transcribes its genome and integrated it into the host cell genome after infection. Figure 1 illustrates the schematic structure of HIV virus (Source: Wikipedia). | |
<center>{{SUSTC-Image|wiki/images/9/9a/SUSTC-Shenzhen-Project-HIV.png}}</center> | <center>{{SUSTC-Image|wiki/images/9/9a/SUSTC-Shenzhen-Project-HIV.png}}</center> | ||
| Line 32: | Line 32: | ||
==CRISPR/Cas system== | ==CRISPR/Cas system== | ||
| - | CRISPR/Cas (Clustered regularly interspaced short palindromic repeats/CRISPR-associated system) is originally a self-protecting mechanism in bacteria against external DNA such as virus genome or plasmids. It uses CAS complex to cut the external viral or plasmid DNA and integrating a short DNA segment into the CRISPR loci in the bacteria genome. This short DNA segment will be transcribed into pre-crRNA and bind to another CAS endonuclease called Cas9 with the help of tracrRNA. The Cas9 endonuclease cuts the external DNA in the existence of not only crRNA but also a special recognizing sequence at the 3’ end of the target DNA called PAM (Protospacer Adjacent Motif). When the same external DNA invades next time, the Cas9-tracrRNA-crRNA complex will recognize and cut it in order to destroy its biological activity. | + | '''CRISPR/Cas''' (Clustered regularly interspaced short palindromic repeats/CRISPR-associated system) is originally a self-protecting mechanism in bacteria against external DNA such as virus genome or plasmids. It uses '''CAS complex''' to '''cut the external viral or plasmid DNA''' and integrating a short DNA segment into the CRISPR loci in the bacteria genome. This short DNA segment will be transcribed into pre-crRNA and bind to another CAS endonuclease called Cas9 with the help of tracrRNA. The Cas9 endonuclease cuts the external DNA in the existence of not only crRNA but also a special recognizing sequence at the 3’ end of the target DNA called PAM (Protospacer Adjacent Motif). When the same external DNA invades next time, the Cas9-tracrRNA-crRNA complex will recognize and cut it in order to destroy its biological activity. |
Figure 2 illustrates the mechanism of Type II CRISPR/Cas system in bacteria (Mali, Esvelt, & Church, 2013). The laboratories of Dr. George Church (Mali, Yang, et al., 2013) and Dr. Feng Zhang (Cong et al., 2013) have successfully developed the Type II CRISPR-Cas system for human cells, which lay the foundation for this project. | Figure 2 illustrates the mechanism of Type II CRISPR/Cas system in bacteria (Mali, Esvelt, & Church, 2013). The laboratories of Dr. George Church (Mali, Yang, et al., 2013) and Dr. Feng Zhang (Cong et al., 2013) have successfully developed the Type II CRISPR-Cas system for human cells, which lay the foundation for this project. | ||
| Line 40: | Line 40: | ||
==PiggyBac (PB) transposon system== | ==PiggyBac (PB) transposon system== | ||
| - | PiggyBac (PB) transposon is a mobile genetic element that efficiently transposes between vectors and chromosomes via a "cut and paste" mechanism but leave no “footprint”. PB transposase in this system recognizes the transposon-specific inverted terminal repeat sequence (PB5 & PB3 in Figure 5) located on both end of transposon vector and efficiently moves the contents from the original sites and efficiently integrates them into TTAA chromosomal sites. Figure 4 illustrates the working mechanism of PB transposon system (Source: Wikipedia; Author: Transposagenbio). | + | '''PiggyBac (PB) transposon''' is a mobile genetic element that '''efficiently transposes between vectors and chromosomes''' via a '''"cut and paste"''' mechanism but leave no '''“footprint”'''. PB transposase in this system recognizes the transposon-specific inverted terminal repeat sequence (PB5 & PB3 in Figure 5) located on both end of transposon vector and efficiently moves the contents from the original sites and efficiently integrates them into TTAA chromosomal sites. Figure 4 illustrates the working mechanism of PB transposon system (Source: Wikipedia; Author: Transposagenbio). |
<center>{{SUSTC-Image|wiki/images/0/06/SUSTC-Shenzhen-Project-PiggyBac-transposon.png}}</center> | <center>{{SUSTC-Image|wiki/images/0/06/SUSTC-Shenzhen-Project-PiggyBac-transposon.png}}</center> | ||
<center>Figure 3. Mechanism of PiggyBac transposon system</center> | <center>Figure 3. Mechanism of PiggyBac transposon system</center> | ||
==A-B toxin== | ==A-B toxin== | ||
| - | A-B | + | A-B toxinare two-component exotoxin secreted by a number of pathogenic bacteria. The complexes contain two subunits called A and B. A subunit is the active portion that is poisonous to host cells. B subunit has a translocation domain and cell receptor binding domain, which '''helps the A subunit going into the cell through endosomes'''. Figure 4 shows how Anthrax toxin (a kind of A-B toxin) enters into human cells (Source: Wikipedia; Author: Y tambe). |
| + | |||
| + | https://2014.igem.org/Team:SUSTC-Shenzhen/Notebook/HeLaCell/Stably_transfect_cell_with_target-sequence_EGFP | ||
| + | |||
<center>{{SUSTC-Image|wiki/images/4/49/SUSTC-Shenzhen-Project-A-B_toxin.png}}</center> | <center>{{SUSTC-Image|wiki/images/4/49/SUSTC-Shenzhen-Project-A-B_toxin.png}}</center> | ||
| Line 54: | Line 57: | ||
<a href="/Team:SUSTC-Shenzhen/Project/Plasmid" class="btn btn-success btn-lg" style="color:#fff">See the full list of plasmids we used.</a> | <a href="/Team:SUSTC-Shenzhen/Project/Plasmid" class="btn btn-success btn-lg" style="color:#fff">See the full list of plasmids we used.</a> | ||
</html> | </html> | ||
| - | ==Constructing EGFP-Cas9 expressing cell | + | ==Constructing EGFP-Cas9 expressing cell lines== |
| - | The program aims to endow the human cell the capacity of resisting the infection of HIV by introducing the CRISPR/Cas9 system into the human cell. To test the efficiency of the Cas9 and explore the optimal conditions of the system, we need a reporter to indicate whether the Cas9 functions and the targeted sites are cut off and destructed. So a cell line integrated with EGFP and Cas9 gene is firstly constructed in a similar way. We use PiggyBac transposon to transfect Hela cell for permanently expressing Cas9 protein. Once the proper gRNA is delivered into these cells, Cas9 binds to the gRNA and start to cut the complementary DNA strand. The vector design is based on Feng Zhang lab’s and Wei Huang lab’s plasmids. | + | The program aims to endow the human cell the capacity of resisting the infection of HIV by '''introducing the CRISPR/Cas9 system into the human cell'''. To test the efficiency of the Cas9 and explore the optimal conditions of the system, we need a reporter to indicate whether the Cas9 functions and the targeted sites are cut off and destructed. So a cell line integrated with EGFP and Cas9 gene is firstly constructed in a similar way. We use PiggyBac transposon to transfect Hela cell for '''permanently expressing Cas9 protein'''. Once the proper gRNA is delivered into these cells, Cas9 binds to the gRNA and start to cut the complementary DNA strand. The vector design is based on Feng Zhang lab’s and Wei Huang lab’s plasmids. |
<center>{{SUSTC-Image|wiki/images/1/17/SUSTC-Shenzhen-Project-PB-TetON-Cas9.jpg}}</center> | <center>{{SUSTC-Image|wiki/images/1/17/SUSTC-Shenzhen-Project-PB-TetON-Cas9.jpg}}</center> | ||
| Line 69: | Line 72: | ||
==Transfecting the cell line with gRNA-encoding plasmid== | ==Transfecting the cell line with gRNA-encoding plasmid== | ||
| - | Cas9 uses gRNA, a 23nt short RNA fragment which binds to its complementary target and 'guide' Cas9 to cut the target DNA. We first check the efficiency of this system | + | Cas9 uses gRNA, a 23nt short RNA fragment which binds to its complementary target and 'guide' Cas9 to cut the target DNA. We first check the efficiency of this system. We construct a plasmid based on Zhang Feng Lab’s pX330 plasmid that encodes a chimeric guide RNA scaffold and mCherry reporter gene followed by a NLS (nucleus leading sequence). It also contains 0, 2, 5 or 7 repeats of UAS sequences which are necessary for the vehicle’s recognition and binding (See Future applications – A-B toxin based gRNA shuttle for details). |
<center>{{SUSTC-Image|wiki/images/b/b2/SUSTC-Shenzhen-Project-gRNA-mCherry-UAS-backbone.jpg}}</center> | <center>{{SUSTC-Image|wiki/images/b/b2/SUSTC-Shenzhen-Project-gRNA-mCherry-UAS-backbone.jpg}}</center> | ||
| Line 79: | Line 82: | ||
<center>Figure 8. Mechanism of gRNA insertion into pX330-derived plasmid backbone</center> | <center>Figure 8. Mechanism of gRNA insertion into pX330-derived plasmid backbone</center> | ||
| - | In this way, with the ratio of reduced green fluorescence and the quantity of red fluorescence we can roughly obtain the efficiency of the CRISPR-Cas system we created. And finally, we reconstruct the plasmid but this time with the | + | In this way, with the ratio of reduced green fluorescence and the quantity of red fluorescence we can roughly obtain the efficiency of the CRISPR-Cas system we created. And finally, we reconstruct the plasmid but this time with the gRNA targeting HIV and test its efficacy. |
<html> | <html> | ||
| - | <a href="/Team:SUSTC-Shenzhen/Project/gRNA-mCherry-UAS_Carried_Plasmids_Design_and_Construction" class="btn btn-success btn-lg" style="color:#fff"> | + | <a href="/Team:SUSTC-Shenzhen/Project/gRNA-mCherry-UAS_Carried_Plasmids_Design_and_Construction" class="btn btn-success btn-lg" style="color:#fff">See how we design and construct our powerful gRNA-mCherry-UAS plasmids</a> |
</html> | </html> | ||
==Deliver gRNA plasmids by A-B toxin-based shuttle== | ==Deliver gRNA plasmids by A-B toxin-based shuttle== | ||
| - | CRISPR/Cas system needs two essential parts for normal function: Cas9 and gRNA. | + | CRISPR/Cas system needs two essential parts for normal function: Cas9 and gRNA. Traditional methods usually transfect the cell with Cas9 and gRNA together (Hu et al., 2014). However, this limits the usage of this system because HIV has high mutated rate, which needs to update gRNA. So we decided to use a flexible nucleic acid delivery system. |
| - | We choose a non-viral DNA delivery system which is based on modified A-B toxin. The chimeric fusion protein mainly comprises 3 parts: target cell-specific binding domain, a translocation domain and a nucleic acid binding domain. The target cell-specific binding domain recognizes the EGF (epidermal growth factor) receptors on the cell surface. The translocation domain enhances nucleic acid escape from the cellular vesicle system and thus to augment nucleic acid transfer. The nucleic acid binding domain, which derives from the yeast GAL4 transcription factor, can carry plasmids with UAS (Upstream Activation Sequence) sequences into cells in vivo. Several research groups | + | We choose a non-viral DNA delivery system which is based on a modified A-B toxin. The chimeric fusion protein mainly comprises 3 parts: target cell-specific binding domain, a translocation domain and a nucleic acid binding domain. The target cell-specific binding domain recognizes the EGF (epidermal growth factor) receptors on the cell surface. The translocation domain enhances nucleic acid escape from the cellular vesicle system and thus to augment nucleic acid transfer. The nucleic acid binding domain, which derives from the yeast GAL4 transcription factor, can carry plasmids with UAS (Upstream Activation Sequence) sequences into cells in vivo. Several research groups (Gaur, Gupta, Goyal, Wels, & Singh, 2002) (Chen et al., 2000) (Fominaya, Uherek, & Wels, 1998) have achieved this goal. Prof. Wels was kindly enough to provide his TEG vehicle for us to deliver DNA into Hela for this project. Figure 7 shows the schematic representation of the TEG fusion gene in the E.coli expression plasmid pWF47-TEG (Wels, Winfried 63110 Rodgau (DE)). A-B toxin delivery system for various human immune cells will need to be developed in the future. |
| + | |||
<center>{{SUSTC-Image|wiki/images/b/b5/SUSTC-Shenzhen-Project-TEG_fusion_protein.png}}</center> | <center>{{SUSTC-Image|wiki/images/b/b5/SUSTC-Shenzhen-Project-TEG_fusion_protein.png}}</center> | ||
| Line 96: | Line 100: | ||
<html> | <html> | ||
| - | <a href="/Team:SUSTC-Shenzhen/Project/A-B_toxin" class="btn btn-success btn-lg" style="color:#fff"> | + | <a href="/Team:SUSTC-Shenzhen/Project/A-B_toxin" class="btn btn-success btn-lg" style="color:#fff">See how we get our A-B toxin-based shuttle</a> |
</html> | </html> | ||
=gRNA design= | =gRNA design= | ||
| - | gRNA is a 23nt short RNA fragment (not include tracrRNA) which binds to its complementary target and 'guide' Cas9 to cut the target DNA. Since we would like to destroy the HIV virus inside human body, a gRNA sequence that matched | + | gRNA is a 23nt short RNA fragment (not include tracrRNA) which binds to its complementary target and 'guide' Cas9 to cut the target DNA. Since we would like to destroy the HIV virus inside human body, a gRNA sequence that matched part of HIV conserved region but have few off-target matches in the human genome is desired. However, designing a gRNA for a human-infective virus is difficult due to the very large difference in the genome size. For virus like HIV, it is even more difficult because HIV can mutate in a very high rate. So we change our goal to finding a group of specific quasi-conservative sequences (gRNA) which are able to target one or more species of HIV. |
| - | Our search for the sequence roughly followed the process described in George M. Church's paper (Mali, Yang, et al., 2013). We modified it slightly to fit our own purpose. We first used bioinformatics method to find the quasi-conservative regions in the HIV-1 whole genome reference library by NIH. From the candidates, we selected the region around 720bp from beginning of the genome (aligned), in the less-selected region slightly off the LTR. | + | Our search for the sequence roughly followed the process described in George M. Church's paper (Mali, Yang, et al., 2013). We modified it slightly to fit our own purpose. We first used bioinformatics method to find the quasi-conservative regions in the HIV-1 whole genome reference library by NIH. From the candidates, we selected the region around 720bp from beginning of the genome (aligned), in the less-selected region slightly off the LTR. We then used the online tools of Feng Zhang's Lab at MIT to find our desired gRNA composition. We also did some calculation based on phenomenological energy calculation to estimate the stability and effectiveness of our gRNA sequences (Hsu et al., 2013). The tool shows that almost no off-target binding will occur. BLAST was used to further confirm the results. We also analyzed the structure of the resulted gRNA, which shows an approximate free energy of -1.4kJ (Zuker, 2003). For detailed information, see our gRNA design page. |
| - | |||
<html> | <html> | ||
| - | <a href="/Team:SUSTC-Shenzhen/gRNA_Design" class="btn btn-success btn-lg" style="color:#fff"> | + | <a href="/Team:SUSTC-Shenzhen/gRNA_Design" class="btn btn-success btn-lg" style="color:#fff">See how we design our gRNA.</a> |
</html> | </html> | ||
| Line 119: | Line 122: | ||
<html> | <html> | ||
| - | <a href="/Team:SUSTC-Shenzhen/Modeling" class="btn btn-success btn-lg" style="color:#fff"> | + | <a href="/Team:SUSTC-Shenzhen/Modeling" class="btn btn-success btn-lg" style="color:#fff">See how we do the modeling.</a> |
</html> | </html> | ||
| Line 126: | Line 129: | ||
<html> | <html> | ||
| - | <a href="/Team:SUSTC-Shenzhen/Safety" class="btn btn-success btn-lg" style="color:#fff"> | + | <a href="/Team:SUSTC-Shenzhen/Safety" class="btn btn-success btn-lg" style="color:#fff">See how we concerned about the Safety.</a> |
</html> | </html> | ||
=Future Perspectives= | =Future Perspectives= | ||
| - | ==In vivo | + | ==In vivo establishment and validation of the system== |
| - | Currently all our experiment design is based on in vitro conditions. However, our ultimate goal is to fight against HIV in vivo. A recent published paper in PNAS | + | Currently all our experiment design is based on in vitro conditions. However, our ultimate goal is to fight against HIV in vivo. A recent published paper in PNAS, sharing the similar design with our project, presented strong evidence for the effect of this system in other cell lines (Hu et al., 2014). So one of our important further works is to establish an in vivo system to test and improve our design. We plan to transplant the engineered Hela cell to immune deficient mice such as Nude or NOD/SCID mice. We will then optimize the dynamics of inducing of Cas9 of the transplanted cell by feeding the mice with Dox. The effect can be monitored in live mice in real time is we conjugate a luciferase gene to Cas9 gene and imaging its activate in the live mice. We will then optimize the delivery method and dosage of the A-B toxin-gRNA plasmid complex and monitor the changing in GFP expression of transplant cell using either biopsy or intravital fluorescent imaging. |
==Permanent implanting Cas9 gene into hematopoietic stem cells== | ==Permanent implanting Cas9 gene into hematopoietic stem cells== | ||
| - | Hematopoietic stem cells are the progenitor of all blood cells, including CD4+ T/B cells. It’s very difficult to extract all CD4+ T/B cells from the human blood system, doing the gene modification and then sent them back. Also, these cells can’t replicate, which means regularly gene modification should be done, which is unrealistic. Instead, one of our team members called Lin Le suggested to modify hematopoietic stem cells because all the offspring of the modified hematopoietic stem cells, including CD4+ T/B cells, carries our system, and it won’t loss since the renewal of CD4+ T/B cell will continue if the modified hematopoietic stem cells are still alive. A recent report (Holt et al., 2010) using engineered | + | Hematopoietic stem cells are the progenitor of all blood cells, including CD4+ T/B cells. It’s very difficult to extract all CD4+ T/B cells from the human blood system, doing the gene modification and then sent them back. Also, these cells can’t replicate, which means regularly gene modification should be done, which is unrealistic. Instead, one of our team members called Lin Le suggested to modify hematopoietic stem cells because all the offspring of the modified hematopoietic stem cells, including CD4+ T/B cells, carries our system, and it won’t loss since the renewal of CD4+ T/B cell will continue if the modified hematopoietic stem cells are still alive. The field of gene therapy has help accumulated a lot of knowledge in this area but it was partially hindered by the viral vectors used. The recent developed high efficient genome editing method including CRISPR/Cas9, Transcription activator-like effector nuclease(TALEN) and zinc-finger nucleases (ZFNs) might provide a safer approach. A recent report (Holt et al., 2010) using engineered ZFNs to eliminate CCR5 in human CD34+ hematopoietic stem /progenitor cells (HSPCs) revealed the possibility to manipulate hematopoietic stem cells. Mouse model used in the report shows positive results after genome editing by ZFNs, indicating promising future for CRISPR/Cas9 mediated genome editing. |
==Using CRISPR/Cas system to integrate Cas9 into human cells== | ==Using CRISPR/Cas system to integrate Cas9 into human cells== | ||
| - | Our current | + | Our current project uses Piggybac transposon system to deliver Cas9 into human cells for proof-of-principle studies. However, the integration site of Piggybac is non-specific, and it also has preferences for active transcribing sites, which may be normal or oncogene sites. Thus it is not ideal for clinical usage. However, the genome editing tool we used in this project (CRISPR/Cas system) has strong specificity for its target and the target can be designed to non-coding regions on human genome by carefully designed gRNA. One of the possible solutions is to integrate homologous recombinant sites around Cas9. By transient transfect the cell with the Cas9 encoding plasmid shown in Figure 13 and the gRNA expression plasmid shown in Figure 7 and use doxycycline to induce the expression of Cas9, we expect to insert our system into the locus which does no harm to normal gene functions. |
<center>{{SUSTC-Image|wiki/images/0/09/SUSTC-Shenzhen-Project-CRISPR-TetON-Cas9.jpg}}</center> | <center>{{SUSTC-Image|wiki/images/0/09/SUSTC-Shenzhen-Project-CRISPR-TetON-Cas9.jpg}}</center> | ||
<center>Figure 13. Possible design of CRISPR-based Cas9 integration plasmid.</center> | <center>Figure 13. Possible design of CRISPR-based Cas9 integration plasmid.</center> | ||
==Using HIV-modified vector to specifically deliver Cas9 gene or gRNA plasmid into CD4+ cells== | ==Using HIV-modified vector to specifically deliver Cas9 gene or gRNA plasmid into CD4+ cells== | ||
| - | One of the main | + | One of the main limitations of our transfection system is non-directional, especially for A-B toxin based gRNA delivery because its target receptor is widely distributed on many cell types. However, the main group our system intends to protect is CD4+ cells because they are most susceptible to HIV. Interestingly, a paper published in 2009 reported a clinical trial that 2 X-linked adrenoleukodystrophy (ALD) patients received very positive treatment effect after gene therapy by using HIV-based lentivirus vector to transfect hematopoietic stem/progenitor cells (HSPCs) ex vivo (Cartier et al., 2009). So we will utilize the targeting capability of lentiviral vector while disarm its genome integration function to enable transient expression of the gRNA in the proper cells in vivo without the side effects of random genome insertion. There’s a saying in Chinese which is ‘Like cures like’. We think it explicitly expressed the idea of this approach. |
==Application in other retroviral diseases== | ==Application in other retroviral diseases== | ||
| - | The system designed by us can also be applied in treating other retrovirus diseases, such as Hepatitis B. We’ve designed gRNA for HBV and constructed target sequence plasmids to test its effectiveness | + | The system designed by us can also be applied in treating other retrovirus diseases, such as Hepatitis B. We’ve designed gRNA for HBV and constructed target sequence plasmids to test its effectiveness. |
| + | |||
| + | <html> | ||
| + | <a href="/Team:SUSTC-Shenzhen/gRNA_Design" class="btn btn-success btn-lg" style="color:#fff">See how we design gRNA for HBV</a> | ||
| + | </html> | ||
=References= | =References= | ||
Latest revision as of 03:59, 18 October 2014
Project Description
a small overview to the whole big ideas
Introduction
In this project, we intended to establish a synthetic biology-based effective HIV-curing system with less side effects. The key goal is to integrate CRISPR/Cas system into human blood system to protect CD4+ cells against viral infection. The gRNA is designed to target the relative conserved regions in HIV viral genome and inactivate its biological activity. Since viral vectors seem to be of limited use in gene therapy strategies (e.g., potential pathogenicity), there is in dire need of a simple, efficient system for cell-specific delivery of nucleic acids. By using non-viral DNA delivery system such as A-B-toxin-GAL4 fusion protein, we can deliver plasmids encoding gRNA into the CD4+ cells and readily attack multiple HIV genome sites simultaneously with the most up-to-date knowledge of HIV epidemic.
Background
HIV (Human immunodeficiency virus)
HIV is the trigger of the lethal disease AIDS. It replicates in and kills T cells, weakens human immune system and allows life-threatening opportunistic infection and cancer to thrive. HIV infects vital cells in the human immune system such as helper T cells (especially CD4+ T cells), macrophages, and dendritic cells. As a retrovirus, HIV reverse transcribes its genome and integrated it into the host cell genome after infection. Figure 1 illustrates the schematic structure of HIV virus (Source: Wikipedia).
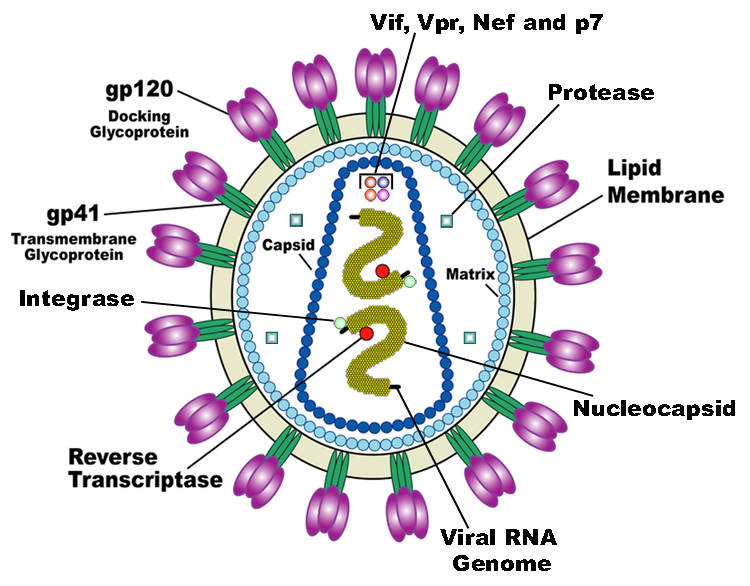
Traditional therapies such as drug treatment can control the development of the disease but not eradicate the virus. The chronic inflammation and immune dysfunction caused by long-term chemotherapy and radiotherapy may also lead to non-AIDS morbidity and mortality (Kiem, Jerome, Deeks, & McCune, 2012). The process of reverse transcription is extremely error-prone, and the resulting mutations may lead to drug resistance or allow the virus to evade the body's immune system.
CRISPR/Cas system
CRISPR/Cas (Clustered regularly interspaced short palindromic repeats/CRISPR-associated system) is originally a self-protecting mechanism in bacteria against external DNA such as virus genome or plasmids. It uses CAS complex to cut the external viral or plasmid DNA and integrating a short DNA segment into the CRISPR loci in the bacteria genome. This short DNA segment will be transcribed into pre-crRNA and bind to another CAS endonuclease called Cas9 with the help of tracrRNA. The Cas9 endonuclease cuts the external DNA in the existence of not only crRNA but also a special recognizing sequence at the 3’ end of the target DNA called PAM (Protospacer Adjacent Motif). When the same external DNA invades next time, the Cas9-tracrRNA-crRNA complex will recognize and cut it in order to destroy its biological activity. Figure 2 illustrates the mechanism of Type II CRISPR/Cas system in bacteria (Mali, Esvelt, & Church, 2013). The laboratories of Dr. George Church (Mali, Yang, et al., 2013) and Dr. Feng Zhang (Cong et al., 2013) have successfully developed the Type II CRISPR-Cas system for human cells, which lay the foundation for this project.
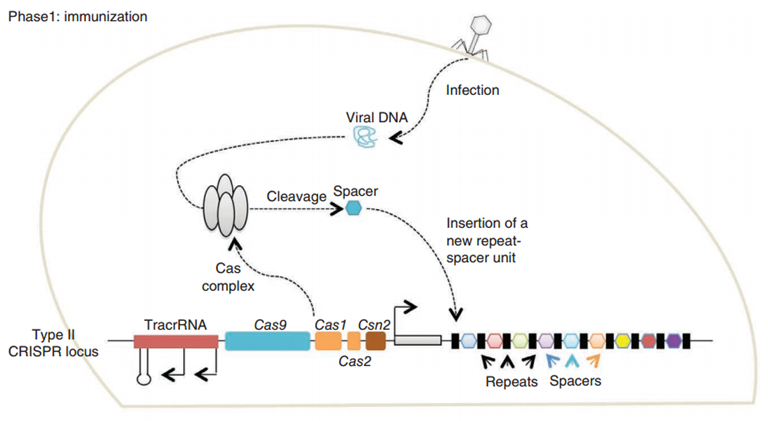
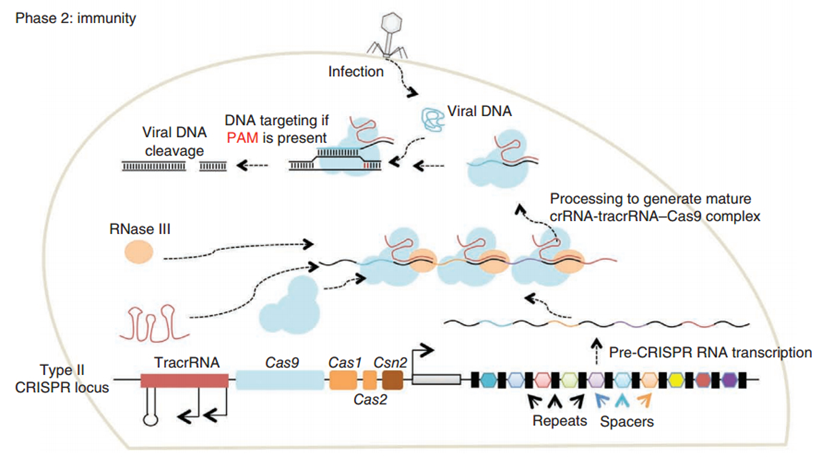
PiggyBac (PB) transposon system
PiggyBac (PB) transposon is a mobile genetic element that efficiently transposes between vectors and chromosomes via a "cut and paste" mechanism but leave no “footprint”. PB transposase in this system recognizes the transposon-specific inverted terminal repeat sequence (PB5 & PB3 in Figure 5) located on both end of transposon vector and efficiently moves the contents from the original sites and efficiently integrates them into TTAA chromosomal sites. Figure 4 illustrates the working mechanism of PB transposon system (Source: Wikipedia; Author: Transposagenbio).
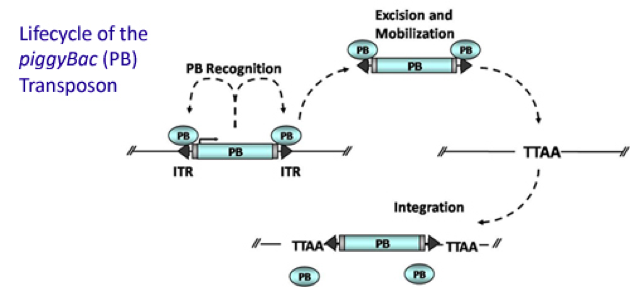
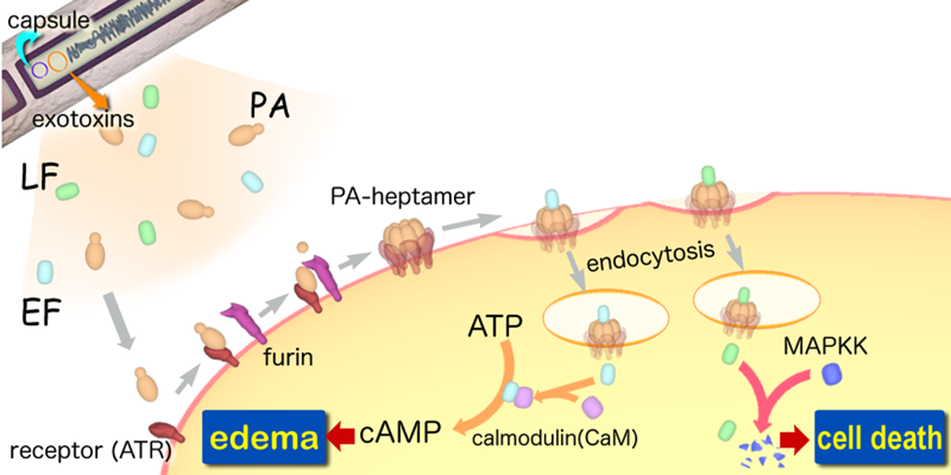
A-B toxin
A-B toxinare two-component exotoxin secreted by a number of pathogenic bacteria. The complexes contain two subunits called A and B. A subunit is the active portion that is poisonous to host cells. B subunit has a translocation domain and cell receptor binding domain, which helps the A subunit going into the cell through endosomes. Figure 4 shows how Anthrax toxin (a kind of A-B toxin) enters into human cells (Source: Wikipedia; Author: Y tambe).
Experiment design
See the full list of plasmids we used.
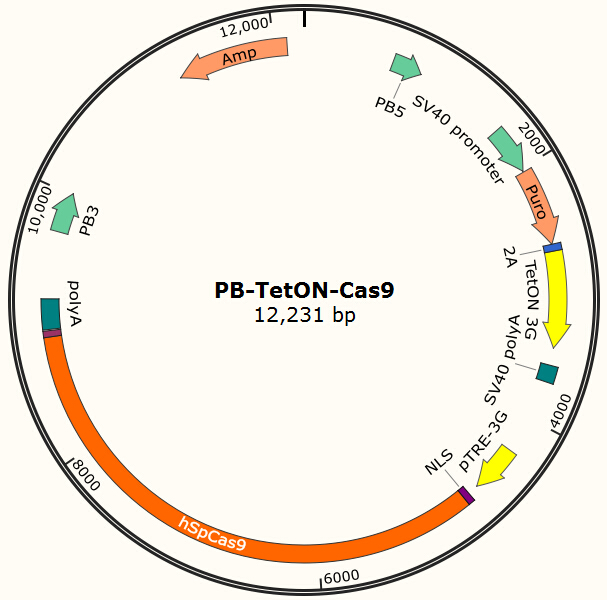
Constructing EGFP-Cas9 expressing cell lines
The program aims to endow the human cell the capacity of resisting the infection of HIV by introducing the CRISPR/Cas9 system into the human cell. To test the efficiency of the Cas9 and explore the optimal conditions of the system, we need a reporter to indicate whether the Cas9 functions and the targeted sites are cut off and destructed. So a cell line integrated with EGFP and Cas9 gene is firstly constructed in a similar way. We use PiggyBac transposon to transfect Hela cell for permanently expressing Cas9 protein. Once the proper gRNA is delivered into these cells, Cas9 binds to the gRNA and start to cut the complementary DNA strand. The vector design is based on Feng Zhang lab’s and Wei Huang lab’s plasmids.
Result Page for Cell Line Construction
Transfecting the cell line with gRNA-encoding plasmid
Cas9 uses gRNA, a 23nt short RNA fragment which binds to its complementary target and 'guide' Cas9 to cut the target DNA. We first check the efficiency of this system. We construct a plasmid based on Zhang Feng Lab’s pX330 plasmid that encodes a chimeric guide RNA scaffold and mCherry reporter gene followed by a NLS (nucleus leading sequence). It also contains 0, 2, 5 or 7 repeats of UAS sequences which are necessary for the vehicle’s recognition and binding (See Future applications – A-B toxin based gRNA shuttle for details).
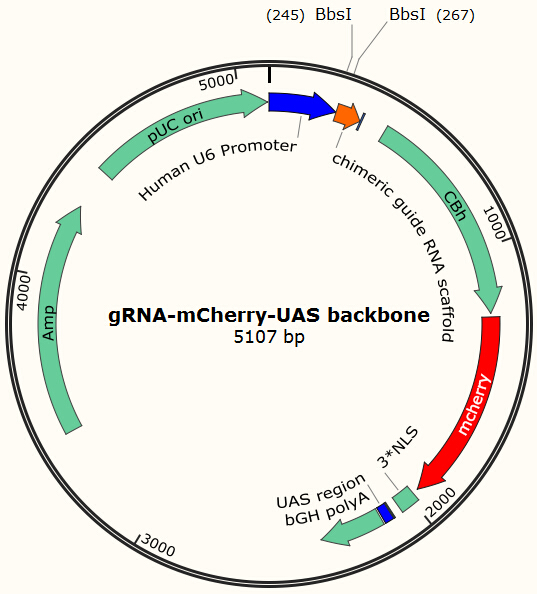
The chimeric guide RNA scaffold is used to insert gRNA. We acquired the gRNA for EGFP from Wei Huang’s lab and put it into our constructed plasmid. Figure 8 shows the design of chimeric guide RNA scaffold (Cong et al., 2013).
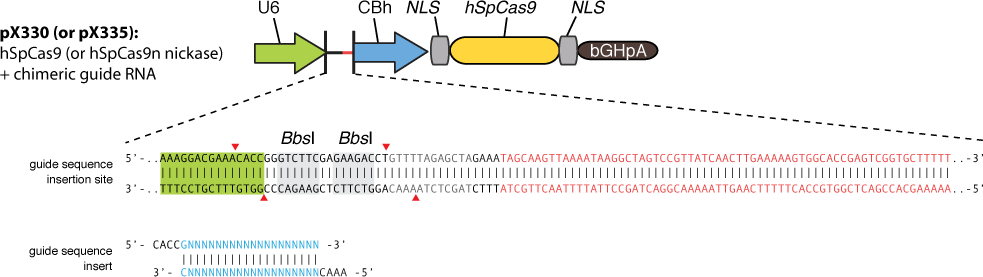
In this way, with the ratio of reduced green fluorescence and the quantity of red fluorescence we can roughly obtain the efficiency of the CRISPR-Cas system we created. And finally, we reconstruct the plasmid but this time with the gRNA targeting HIV and test its efficacy.
See how we design and construct our powerful gRNA-mCherry-UAS plasmids
Deliver gRNA plasmids by A-B toxin-based shuttle
CRISPR/Cas system needs two essential parts for normal function: Cas9 and gRNA. Traditional methods usually transfect the cell with Cas9 and gRNA together (Hu et al., 2014). However, this limits the usage of this system because HIV has high mutated rate, which needs to update gRNA. So we decided to use a flexible nucleic acid delivery system. We choose a non-viral DNA delivery system which is based on a modified A-B toxin. The chimeric fusion protein mainly comprises 3 parts: target cell-specific binding domain, a translocation domain and a nucleic acid binding domain. The target cell-specific binding domain recognizes the EGF (epidermal growth factor) receptors on the cell surface. The translocation domain enhances nucleic acid escape from the cellular vesicle system and thus to augment nucleic acid transfer. The nucleic acid binding domain, which derives from the yeast GAL4 transcription factor, can carry plasmids with UAS (Upstream Activation Sequence) sequences into cells in vivo. Several research groups (Gaur, Gupta, Goyal, Wels, & Singh, 2002) (Chen et al., 2000) (Fominaya, Uherek, & Wels, 1998) have achieved this goal. Prof. Wels was kindly enough to provide his TEG vehicle for us to deliver DNA into Hela for this project. Figure 7 shows the schematic representation of the TEG fusion gene in the E.coli expression plasmid pWF47-TEG (Wels, Winfried 63110 Rodgau (DE)). A-B toxin delivery system for various human immune cells will need to be developed in the future.
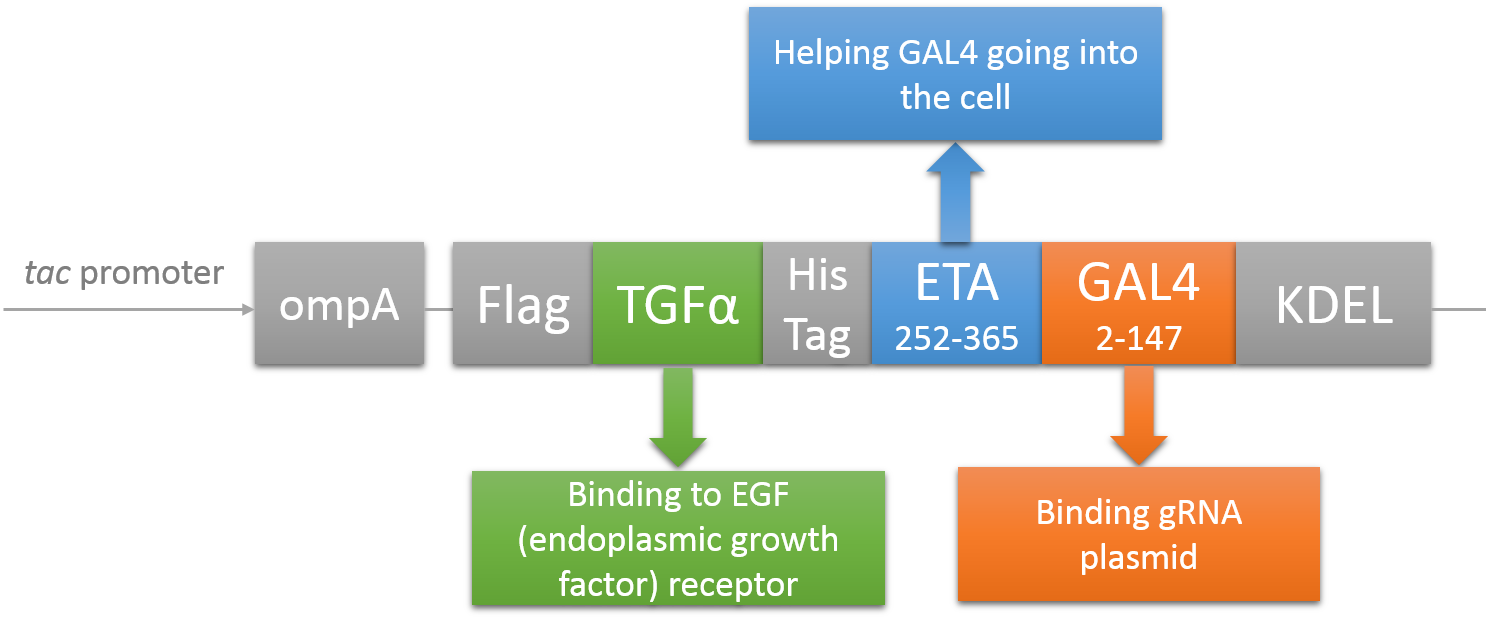
See how we get our A-B toxin-based shuttle
gRNA design
gRNA is a 23nt short RNA fragment (not include tracrRNA) which binds to its complementary target and 'guide' Cas9 to cut the target DNA. Since we would like to destroy the HIV virus inside human body, a gRNA sequence that matched part of HIV conserved region but have few off-target matches in the human genome is desired. However, designing a gRNA for a human-infective virus is difficult due to the very large difference in the genome size. For virus like HIV, it is even more difficult because HIV can mutate in a very high rate. So we change our goal to finding a group of specific quasi-conservative sequences (gRNA) which are able to target one or more species of HIV.
Our search for the sequence roughly followed the process described in George M. Church's paper (Mali, Yang, et al., 2013). We modified it slightly to fit our own purpose. We first used bioinformatics method to find the quasi-conservative regions in the HIV-1 whole genome reference library by NIH. From the candidates, we selected the region around 720bp from beginning of the genome (aligned), in the less-selected region slightly off the LTR. We then used the online tools of Feng Zhang's Lab at MIT to find our desired gRNA composition. We also did some calculation based on phenomenological energy calculation to estimate the stability and effectiveness of our gRNA sequences (Hsu et al., 2013). The tool shows that almost no off-target binding will occur. BLAST was used to further confirm the results. We also analyzed the structure of the resulted gRNA, which shows an approximate free energy of -1.4kJ (Zuker, 2003). For detailed information, see our gRNA design page.
Modeling
Our project is intended to treat retrovirus diseases.So we want to know how effective the CRISPR/Cas9 system is. In our model, we discuss about dynamic changes of different cells in a person’s body using Matlab and we take HIV for exmple. After theoretical analysis, symbol description, formula derivation and Matlab analysis, we can see how the system works and the influence the CRISPR/Cas system produces by the change of virus and cells in this model. It will help us to forecast the future applications of our project.If the environment doesn't change, we can stimulate the situation in Matlab for a short time. The result is that the CRISPR/Cas9 system will take effect.
Safety
Our system has several built-in design to reduce the potential safety risks of the system utilization.
See how we concerned about the Safety.
Future Perspectives
In vivo establishment and validation of the system
Currently all our experiment design is based on in vitro conditions. However, our ultimate goal is to fight against HIV in vivo. A recent published paper in PNAS, sharing the similar design with our project, presented strong evidence for the effect of this system in other cell lines (Hu et al., 2014). So one of our important further works is to establish an in vivo system to test and improve our design. We plan to transplant the engineered Hela cell to immune deficient mice such as Nude or NOD/SCID mice. We will then optimize the dynamics of inducing of Cas9 of the transplanted cell by feeding the mice with Dox. The effect can be monitored in live mice in real time is we conjugate a luciferase gene to Cas9 gene and imaging its activate in the live mice. We will then optimize the delivery method and dosage of the A-B toxin-gRNA plasmid complex and monitor the changing in GFP expression of transplant cell using either biopsy or intravital fluorescent imaging.
Permanent implanting Cas9 gene into hematopoietic stem cells
Hematopoietic stem cells are the progenitor of all blood cells, including CD4+ T/B cells. It’s very difficult to extract all CD4+ T/B cells from the human blood system, doing the gene modification and then sent them back. Also, these cells can’t replicate, which means regularly gene modification should be done, which is unrealistic. Instead, one of our team members called Lin Le suggested to modify hematopoietic stem cells because all the offspring of the modified hematopoietic stem cells, including CD4+ T/B cells, carries our system, and it won’t loss since the renewal of CD4+ T/B cell will continue if the modified hematopoietic stem cells are still alive. The field of gene therapy has help accumulated a lot of knowledge in this area but it was partially hindered by the viral vectors used. The recent developed high efficient genome editing method including CRISPR/Cas9, Transcription activator-like effector nuclease(TALEN) and zinc-finger nucleases (ZFNs) might provide a safer approach. A recent report (Holt et al., 2010) using engineered ZFNs to eliminate CCR5 in human CD34+ hematopoietic stem /progenitor cells (HSPCs) revealed the possibility to manipulate hematopoietic stem cells. Mouse model used in the report shows positive results after genome editing by ZFNs, indicating promising future for CRISPR/Cas9 mediated genome editing.
Using CRISPR/Cas system to integrate Cas9 into human cells
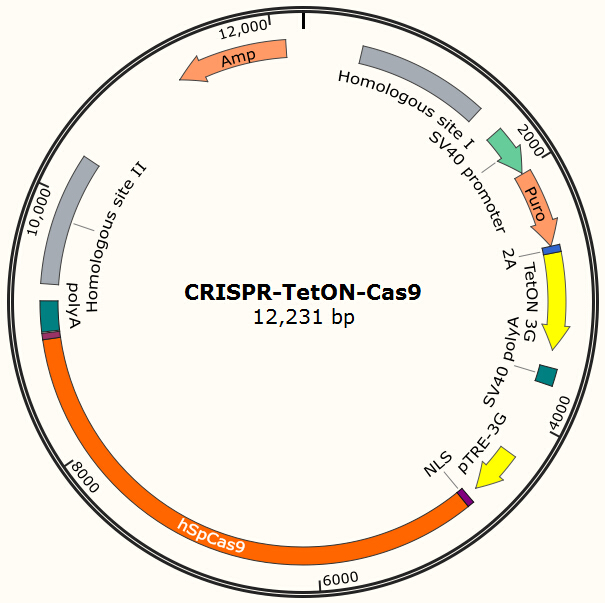
Our current project uses Piggybac transposon system to deliver Cas9 into human cells for proof-of-principle studies. However, the integration site of Piggybac is non-specific, and it also has preferences for active transcribing sites, which may be normal or oncogene sites. Thus it is not ideal for clinical usage. However, the genome editing tool we used in this project (CRISPR/Cas system) has strong specificity for its target and the target can be designed to non-coding regions on human genome by carefully designed gRNA. One of the possible solutions is to integrate homologous recombinant sites around Cas9. By transient transfect the cell with the Cas9 encoding plasmid shown in Figure 13 and the gRNA expression plasmid shown in Figure 7 and use doxycycline to induce the expression of Cas9, we expect to insert our system into the locus which does no harm to normal gene functions.
Using HIV-modified vector to specifically deliver Cas9 gene or gRNA plasmid into CD4+ cells
One of the main limitations of our transfection system is non-directional, especially for A-B toxin based gRNA delivery because its target receptor is widely distributed on many cell types. However, the main group our system intends to protect is CD4+ cells because they are most susceptible to HIV. Interestingly, a paper published in 2009 reported a clinical trial that 2 X-linked adrenoleukodystrophy (ALD) patients received very positive treatment effect after gene therapy by using HIV-based lentivirus vector to transfect hematopoietic stem/progenitor cells (HSPCs) ex vivo (Cartier et al., 2009). So we will utilize the targeting capability of lentiviral vector while disarm its genome integration function to enable transient expression of the gRNA in the proper cells in vivo without the side effects of random genome insertion. There’s a saying in Chinese which is ‘Like cures like’. We think it explicitly expressed the idea of this approach.
Application in other retroviral diseases
The system designed by us can also be applied in treating other retrovirus diseases, such as Hepatitis B. We’ve designed gRNA for HBV and constructed target sequence plasmids to test its effectiveness.
See how we design gRNA for HBV
References
Cartier, N., Hacein-Bey-Abina, S., Bartholomae, C. C., Veres, G., Schmidt, M., Kutschera, I., . . . Aubourg, P. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science, 326(5954), 818-823. doi: 10.1126/science.1171242
Chen, T. Y., Hsu, C. T., Chang, K. H., Ting, C. Y., Whang-Peng, J., Hui, C. F., & Hwang, J. (2000). Development of DNA delivery system using Pseudomonas exotoxin A and a DNA binding region of human DNA topoisomerase I. Applied Microbiology and Biotechnology, 53(5), 558-567.
Clontech. Tet-On 3G Inducible Expression Systems User Manual. http://www.clontech.com/TW/Products/Inducible_Systems/Tetracycline-Inducible_Expression/ibcGetAttachment.jsp?cItemId=17569&fileId=6832390&sitex=10021:22372:US.
Cong, L., Ran, F. A., Cox, D., Lin, S. L., Barretto, R., Habib, N., . . . Zhang, F. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science, 339(6121), 819-823. doi: DOI 10.1126/science.1231143
Fominaya, J., Uherek, C., & Wels, W. (1998). A chimeric fusion protein containing transforming growth factor-alpha mediates gene transfer via binding to the EGF receptor. Gene Ther, 5(4), 521-530. doi: 10.1038/sj.gt.3300614
Gaur, R., Gupta, P. K., Goyal, A., Wels, W., & Singh, Y. (2002). Delivery of nucleic acid into mammalian cells by anthrax toxin. Biochemical and Biophysical Research Communications, 297(5), 1121-1127. doi: Pii S0006-291x(02)02299-4 Doi 10.1016/S0006-291x(02)02299-4
Holt, N., Wang, J., Kim, K., Friedman, G., Wang, X., Taupin, V., . . . Cannon, P. M. (2010). Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nature Biotechnology, 28(8), 839-847. doi: 10.1038/nbt.1663
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., . . . Zhang, F. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology, 31(9), 827-+. doi: Doi 10.1038/Nbt.2647
Hu, W., Kaminski, R., Yang, F., Zhang, Y., Cosentino, L., Li, F., . . . Khalili, K. (2014). RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1405186111
Kiem, H. P., Jerome, K. R., Deeks, S. G., & McCune, J. M. (2012). Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell, 10(2), 137-147. doi: 10.1016/j.stem.2011.12.015
Mali, P., Esvelt, K. M., & Church, G. M. (2013). Cas9 as a versatile tool for engineering biology. Nat Methods, 10(10), 957-963. doi: 10.1038/nmeth.2649
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., . . . Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science, 339(6121), 823-826. doi: 10.1126/science.1232033
Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res, 31(13), 3406-3415. doi: Doi 10.1093/Nar/Gkg595