 "
"
Team:Heidelberg/pages/PCR 2.0
From 2014.igem.org
(→Thermal stability of circular mDNMT1s(731-1602)) |
|||
| (183 intermediate revisions not shown) | |||
| Line 1: | Line 1: | ||
<html> | <html> | ||
| - | |||
<div class="panel panel-default"> | <div class="panel panel-default"> | ||
| - | <div class="panel- | + | <div class="panel-body" style="background-color:lightgray; padding:15px"> |
| + | <div class="row"> | ||
| + | <div class="col-md-6"> | ||
| + | <img class="img-responsive" src="https://static.igem.org/mediawiki/2014/8/8c/Dnmt1_GraphicalAbstract.001.png"> | ||
</div> | </div> | ||
| - | <div class= | + | <div class="col-md-6"> |
| - | < | + | <p style="font-size:25px; font-weight:bold">Abstract</p> |
| - | < | + | <br/> |
| + | <p>DNA methylation is the most abundant DNA modification and essential for embryonic development, gene regulation and genomic stability. Although several methods for the detection of methylation patterns exist, there is no easy way to amplify methylated DNA for <i>in vitro</i> or <i>in vivo</i> studies.</p> | ||
| + | <p>To empower epigenetic research, we envisioned a PCR2.0 which maintains DNA methylation patterns during amplification. The central element of this PCR2.0 is a heat-resistant DNA methyltransferase - DNMT1 (731-1602) - which we created by circularization using our intein toolbox and the CRAUT linker software.</p> | ||
| + | <p>So far, our DNMT1 represents the largest circularized protein which highlights the usefulness of our intein toolbox in combination with the CRAUT linker software. Increased heat resistance of our circular DNMT1 which was observed in initial assays smoothens the path for the establishment of a PCR2.0 and illustrates the suitability of intein-mediated circularization for the advancement of heat resistant proteins.</p> | ||
| + | <p style="font-size:25px; font-weight:bold">Highlights</p> | ||
| + | <ul> | ||
| + | <li>circularization seems to increase heat stability of mDNMT1 (731-1602): successful first step towards PCR2.0.</li> | ||
| + | <li>mDNMT1 is the largest circularized protein (105kDa) so far.</li> | ||
| + | <li>efficient expression and purification of active linear and circular mDNMT1 (731-1602) in E.coli.</li> | ||
| + | <li>successful application of CRAUT linker design software and intein toolbox for circularization.</li> | ||
| + | </ul> | ||
</div> | </div> | ||
</div> | </div> | ||
</div> | </div> | ||
| + | </div> | ||
| + | |||
</html> | </html> | ||
| Line 16: | Line 30: | ||
==Motivation== | ==Motivation== | ||
| - | The invention of the polymerase chain reaction in 1983 by Kary | + | The invention of the polymerase chain reaction in 1983 by Kary Mullis revolutionized our world by enabling amplification of DNA in an exponential manner. Further improvements including the use of thermo-stable DNA polymerases made the method even more efficient, allowing its widespread use in nearly every field of modern diagnostics and research. However, a major part of information is lost when using conventional PCR as DNA modifications are not copied. The most abundant modification is DNA methylation, which is a prominent regulator of gene expression in all kingdoms of life. Methylation and other modifications that influence the DNA function without changing its actual sequence are studied in the fast-growing field of epigenetics. Even though the detection and mapping of methylation patterns have become feasible in a high-throughput manner by using bisulfite sequencing and array techniques [[#References|[1]]], further functional analysis is still hindered by the small amount of primary material. Up to now, there is no method available to amplify methylated DNA without knowledge of methylation patterns and expensive de-novo synthesis. |
| - | To empower epigenetics, we propose the | + | To empower epigenetics, we propose the '''“PCR 2.0”''' as an easy and efficient way to amplify DNA templates in an exponential manner while maintaining their specific methylation pattern. The pivotal element of this PCR reaction is a heat-stable DNA methyltransferase (DNMT). Although comparable enzymes exist in thermophile organisms [[#References|[2]]], until now no suitable protein has been found or synthesized that withstands the harsh conditions of a PCR. |
| - | + | To achieve this goal of a PCR2.0 that can be used to amplify DNA including its intrinsic methylation pattern, our main aim was the generation of a heat-stable Dnmt1 that can be produced in a larger scale and shows efficient and specific methylation of DNA. It was reported that the insertion of additional intramolecular bonds can increase the stability of proteins [[#References|[3]]][[#References|[4]]], and that specifically joining the C- and N-terminus improves the thermostability of smaller peptides [[#References|[5]]]. This is why our approach to generate a heat-stable Dnmt1 applies: | |
| + | * protein circularization using our intein toolbox | ||
| + | * creating a potent [https://2014.igem.org/Team:Heidelberg/Software/Linker_Software Software] to design efficient linker peptides connecting C- and N-terminus | ||
| - | To | + | To enable the PCR2.0 we started the circularization of the largest protein so far… |
| - | + | ||
==Epigenetics - there is more than just A, T, C and G…== | ==Epigenetics - there is more than just A, T, C and G…== | ||
| - | In mammals and other vertebrates methylation occurs at cytosine nucleotides that are followed by guanines (CpG) | + | In mammals and other vertebrates DNA methylation occurs at the C5 position of cytosine nucleotides that are followed by guanines (CpG). It is a prominent key regulator of transcription, embryonic development, X chromosome inactivation and many other cellular functions [[#References|[6]]][[#References|[7]]]. The human genome contains about 28 million CpGs of which 60-80% are methylated [[#References|[28]]]. Due to its great prevalence and the heredity of this modification, the 5-methylcytosine (5mC) is also known as the “fifth base” of eukaryotic genomes [[#References|[9]]]. DNA methylation is preferentially occurring at intergenic regions and repetitive sequences, where it is known to silence gene expression [[#References|[10]]][[#References|[11]]][[#References|[12]]]. Inappropriate cytosine methylation and silencing has been reported to play a crucial role in the development of numerous diseases, including cancer, imprinting diseases and repeat-instability based diseases such Huntington's disease [[#References|[13]]]. |
==A closer look at DNA Methyltransferases== | ==A closer look at DNA Methyltransferases== | ||
| Line 36: | Line 51: | ||
file=methylation_principle.png}} | file=methylation_principle.png}} | ||
| - | The family of enzymes called DNA methyltransferases (DNMTs) is responsible for the establishment and maintenance of cell-type specific DNA methylation patterns. Whereas the two enzymes DNMT3a and DNMT3b contribute to de novo methylation of DNA during development, DNMT1 is preserving existing methylation patterns throughout cell divisions. To do so, DNMT1 exploits the principle of | + | |
| + | The family of enzymes called DNA methyltransferases (DNMTs) is responsible for the establishment and maintenance of cell-type specific DNA methylation patterns. Whereas the two enzymes DNMT3a and DNMT3b contribute to de novo methylation of DNA during development, DNMT1 is preserving existing methylation patterns throughout cell divisions. To do so, DNMT1 exploits the principle of semi-conservative replication, using the parental strand as a template to create an exact copy of methylation patterns on the daughter strand (Figure 1). After DNA synthesis, the DNA consists of a methylated and an unmethylated strand. DNMT1 recognizes these so called hemi-methylated CpG sites, and transfers a methyl group from the methyl donor S-Adenosyl-methionine (SAM) to the cytosine belonging to the newly synthesized strand [[#References|[15]]][[#References|[19]]]. | ||
| Line 42: | Line 58: | ||
align=left| | align=left| | ||
caption=Figure 2) Crystal structure of full-length and truncated (731-1602) mDNMT1| | caption=Figure 2) Crystal structure of full-length and truncated (731-1602) mDNMT1| | ||
| - | descr=Structural overview of full length mDNMT1 and truncated mDNMT1(731–1602) (A) Color-coded schematic overview of domain structure and numbering of mDNMT1 sequence. (B) Ribbon presentation of full-length DNMT1 with the CXXC,BAH1, BAH2,and methyltransferase domain colored in light blue, yellow, orange and red, respectively. (C) Crystal structure of truncated DNMT1, missing the CXXC domain and schowing more adjacent protein termini | + | descr=Structural overview of full length mDNMT1 and truncated mDNMT1 (731–1602) (A) Color-coded schematic overview of domain structure and numbering of mDNMT1 sequence. (B) Ribbon presentation of full-length DNMT1 with the CXXC, BAH1, BAH2,and methyltransferase domain colored in light blue, yellow, orange and red, respectively. (C) Crystal structure of truncated DNMT1, missing the CXXC domain and schowing more adjacent protein termini that the full-length version.| |
file=dnmt1_structure.png}} | file=dnmt1_structure.png}} | ||
| - | The complex structure of DNMT1 and several truncated versions have been recently solved by X-ray crystallography [[#References|[19]]]. For our experiments we used the smallest truncated version of DNMT1 that has been reported to efficiently mediate methylation maintenance. This truncated version | + | |
| + | |||
| + | The complex structure of DNMT1 and several truncated versions have been recently solved by X-ray crystallography [[#References|[19]]]. For our experiments we used the smallest truncated version of DNMT1 - mDNMT1 (731-1602) - that has been reported to efficiently mediate methylation maintenance. This truncated version is derived from <i>Mus musculus</i> and comprises the amino acids 731-1602 of the full length protein [[#References|[15]]][[#References|[19]]] (kindly provided by Dr. Bashtrykov after approval from Prof. Patel). Murine DNMT1 (731-1602) contains a C-terminal catalytic methyltransferase domain as well as the bromo-adjacent homology (BAH) domains, which prevent the binding of unmethylated DNA to the catalytic core. The truncated enzyme is missing the CXXC domain, which has a high affinity to hemi-methylated CpG dinucleotides, but was shown to be dispensable for protein function [[#References|[16]]]. In contrast to the full-length DNMT1, mDNMT1 (731-1602) has the great advantage of being efficiently expressed in E.coli. Another benefit of the truncated DNMT1 is that the N- and C- terminus are closer together than in the full-length DNMT1 (Figure 2). Thus circularization might cause less deformation and show lower impact on the overall activity, which makes this truncated mDNMT1 an ideal candidate for our purposes. | ||
| + | |||
| + | <div style="clear:both;"></div> | ||
==Circularization - The missing link== | ==Circularization - The missing link== | ||
| - | Heat stabilization of mDNMT1 as a pivotal element of our methylation-maintaining PCR2.0 was approached by circularization of the protein. Until now, only smaller proteins have been stabilized using this method. Therefore, to our knowledge, mDNMT1 – even in its shortest truncated version of 871 amino acids and a molecular weight of approximately 100kDa – is the largest protein that has ever been tried to circularize. According to the crystal structure, the distance between the termini of the truncated DNMT1 is 48 Å. We suspected that circularization of the protein by direct fusion of both termini might cause deformation of the protein structure. Therefore we used our own [https://2014.igem.org/Team:Heidelberg/Software/Linker_Software CRAUT Linker Software] to create a linking amino acid sequence, adapted to the structure of DNMT1. The software is able to design protein linkers with the required length to bridge the gap between the protein termini while bypassing the catalytic core of the enzyme. | + | Heat stabilization of mDNMT1 as a pivotal element of our methylation-maintaining PCR2.0 was approached by circularization of the protein. Until now, only smaller proteins have been stabilized using this method. Therefore, to our knowledge, mDNMT1 – even in its shortest truncated version of 871 amino acids and a molecular weight of approximately 100kDa – is the largest protein that has ever been tried to circularize. According to the crystal structure, the distance between the termini of the truncated DNMT1 is 48 Å. We suspected that circularization of the protein by direct fusion of both termini might cause deformation of the protein structure. Therefore we used our own [https://2014.igem.org/Team:Heidelberg/Software/Linker_Software CRAUT Linker Software] to create a linking amino acid sequence, adapted to the structure of DNMT1. The software is able to design protein linkers with the required length to bridge the gap between the protein termini while bypassing the catalytic core of the enzyme. |
| - | Since we could not estimate the impact of the linker introduction, we focused on two different kinds of linkers: a so called rigid linker that has been calculated and optimized during the establishment of our linker software and a flexible peptide connection consisting mostly of glycine and serine. | + | Since we could not estimate the impact of the linker introduction, we focused on two different kinds of linkers: a so called rigid linker that has been calculated and optimized during the establishment of our linker software and a flexible peptide connection consisting mostly of glycine and serine. To perform circularization we have used the split NpuDnaE Intein, since it is used as the <i>golden standard</i> for protein splicing and has been efficiently tested by us through circularization of GFP, Lysozyme and Xylanase. Moreover, we tried to implement protein circularization by using sortase A, which is recognizing and cleaving a carboxyterminal sorting signal followed by a transpeptidation reaction that can be exploited for protein circularization. This method has been reported to be very efficient [[#References|[4]]] and was therefore included in our study as possible candidate for large scale productions of circular proteins. |
| - | To perform circularization we have used the split NpuDnaE Intein since it is used as the <i> golden standard </i> | + | |
| - | = | + | =Experimental procedures= |
==Constructs== | ==Constructs== | ||
| - | |||
We designed constructs in order to characterize the efficiency of sortase A as well as intein mediated circularization of DNMT1. Both approaches are based on the comparison of the circularized protein with a corresponding linear counterpart at different temperatures and incubation times. | We designed constructs in order to characterize the efficiency of sortase A as well as intein mediated circularization of DNMT1. Both approaches are based on the comparison of the circularized protein with a corresponding linear counterpart at different temperatures and incubation times. | ||
| - | + | {{:Team:Heidelberg/templates/image-half| | |
| + | align=right| | ||
| + | caption=Construct for circularization of mDNMT1 using inteins.| | ||
| + | descr= | | ||
| + | file=Dnmt1_construct1.png}} | ||
For circularization of DNMT1 with inteins we fused the obtained truncated version of DNMT1 (731-1602) [[#References|[15]]] with an appropriate linker and split NpuDnaE domains at either site of the construct. For efficient purification of the protein, all constructs contained a hexa-histidine tag. Moreover, we have cloned the ubiquitin-like Smt3 protein from <i> Saccharomyces cerevisiae </i> in front of the DNMT1-intein complex. This attachment is used to increase the yield when expressing large mammalian proteins like DNMT1 in <i>E.coli</i> [[#References|[17]]]. The constructs have been designed in a way that Smt3 is no longer included in the final circular protein, and therefore cannot interfere with its function. | For circularization of DNMT1 with inteins we fused the obtained truncated version of DNMT1 (731-1602) [[#References|[15]]] with an appropriate linker and split NpuDnaE domains at either site of the construct. For efficient purification of the protein, all constructs contained a hexa-histidine tag. Moreover, we have cloned the ubiquitin-like Smt3 protein from <i> Saccharomyces cerevisiae </i> in front of the DNMT1-intein complex. This attachment is used to increase the yield when expressing large mammalian proteins like DNMT1 in <i>E.coli</i> [[#References|[17]]]. The constructs have been designed in a way that Smt3 is no longer included in the final circular protein, and therefore cannot interfere with its function. | ||
| - | All cloning steps were carried out using our new RFC | + | All cloning steps were carried out using our new RFC[i] standard procedure. |
| + | |||
The mDNMT1 (731-1602) constructs that have been designed to allow sortase A mediated cirularization are flanked by the sortase A recognition sequence LPETGG on the C–terminus as well as N-terminal glycines that that can be exposed through previous TEV treatment. Since the transpeptidation via sortase requires an additional <i> in vitro </i> reaction, subsequent purification is necessary. To facilitate this step and to enrich the circular product in the sample, a hexa-histidine tag is present in the initial protein that is lost during circularization. Hence, an additional affinity chromatography via His trap can be used to separate the histidine tag containing sortase and not circularized educt from the circular flow-through. | The mDNMT1 (731-1602) constructs that have been designed to allow sortase A mediated cirularization are flanked by the sortase A recognition sequence LPETGG on the C–terminus as well as N-terminal glycines that that can be exposed through previous TEV treatment. Since the transpeptidation via sortase requires an additional <i> in vitro </i> reaction, subsequent purification is necessary. To facilitate this step and to enrich the circular product in the sample, a hexa-histidine tag is present in the initial protein that is lost during circularization. Hence, an additional affinity chromatography via His trap can be used to separate the histidine tag containing sortase and not circularized educt from the circular flow-through. | ||
| Line 82: | Line 105: | ||
align=right| | align=right| | ||
caption=Figure 3) Purification of recombinant mDNMT1| | caption=Figure 3) Purification of recombinant mDNMT1| | ||
| - | descr= The purification of recombinantly expressed mDNMT1 is a challenging task including large scale induction, efficient cell lysis, reduction of sample volume via ultracentrifugation, immobilized Ni | + | descr= The purification of recombinantly expressed mDNMT1 is a challenging task including large scale induction, efficient cell lysis, reduction of sample volume via ultracentrifugation, immobilized Ni ion affinity chromatography, dialysis and concentration through centrifugation. | |
file=Purification_procedure_new.png}} | file=Purification_procedure_new.png}} | ||
| - | In comparison to the full length murine DNMT1, our modified mDNMT1(731-1602) can be expressed in <i>E.coli (Rosetta DE3)</i> and does not need the establishment of Baculovirus based system and the use of insect cell cultures. First experiments were conducted to determine the optimal conditions for protein induction and expression. We tested several concentrations of IPTG as well as different temperatures to maximize the yield of mDNMT1. Still, even though expression of the constructs can be simplified by using <i>E.coli</i>, purification of the recombinant protein remains a challenging task requiring multiple different steps that had to be adapted and established from published protocols [[#References|[19]]]. | + | In comparison to the full length murine DNMT1, our modified mDNMT1 (731-1602) can be expressed in <i>E.coli (Rosetta DE3)</i> and does not need the establishment of Baculovirus based system and the use of insect cell cultures. First experiments were conducted to determine the optimal conditions for protein induction and expression. We tested several concentrations of IPTG as well as different temperatures to maximize the yield of mDNMT1. Still, even though expression of the constructs can be simplified by using <i>E.coli</i>, purification of the recombinant protein remains a challenging task requiring multiple different steps that had to be adapted and established from published protocols [[#References|[19]]]. |
In a first step, we optimized the process of cell lysis, to be able to extract a maximum amount of active protein from our cultures. Therefore we compared the two commonly used methods sonication and disruption via French press. Subsequently, the sample was cleared from cell debris by ultracentrifugation. The protein of interest containing a hexa-histidine tag was further enriched via immobilized metal ion affinity chromatography (IMAC) using a His trap and eluted with imidazol. In a next step, low molecular weight solutes such as salts and imidazol that would interfere with protein function were removed from the sample by dialysis. Finally, the purified protein was concentrated by size-exclusion centrifugation using filters with an appropriate mass cutoff. | In a first step, we optimized the process of cell lysis, to be able to extract a maximum amount of active protein from our cultures. Therefore we compared the two commonly used methods sonication and disruption via French press. Subsequently, the sample was cleared from cell debris by ultracentrifugation. The protein of interest containing a hexa-histidine tag was further enriched via immobilized metal ion affinity chromatography (IMAC) using a His trap and eluted with imidazol. In a next step, low molecular weight solutes such as salts and imidazol that would interfere with protein function were removed from the sample by dialysis. Finally, the purified protein was concentrated by size-exclusion centrifugation using filters with an appropriate mass cutoff. | ||
| Line 99: | Line 122: | ||
file=DNMT1_substrate.png}} | file=DNMT1_substrate.png}} | ||
| - | To measure the efficiency of mDNMT1 maintenance methylation, we used an assay that is based on two methylation-sensitive restriction enzymes Sau3AI and HpaII. It relies on the inhibition of restriction enzymes when methyl groups are attached to the cleavage site. We used a 40-mer DNA template with one hemi-methylated and one unmethylated CpG site which are located within the Sau3AI and HpaII restriction site, respectively (adapted from Bashtrykov et colleagues [[#References|[16]]]. | + | To measure the efficiency and specificity of mDNMT1 maintenance methylation activity, we used an assay that is based on the two methylation-sensitive restriction enzymes Sau3AI and HpaII. It relies on the inhibition of restriction enzymes when methyl groups are attached to the cleavage site. We used a 40-mer DNA template with one hemi-methylated and one unmethylated CpG site which are located within the Sau3AI and HpaII restriction site, respectively (adapted from Bashtrykov et colleagues [[#References|[16]]]. |
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
| Line 107: | Line 130: | ||
file=MethylationAssay_overview.png}} | file=MethylationAssay_overview.png}} | ||
| - | The restriction enzyme Sau3AI is capable of cleaving the template despite of the hemimethylated state of the first CpG nucleotide. Similarly, HpaII can attack its unmethylated native site, leading to fragmentation of the template as long as no further methylation occurs | + | The restriction enzyme Sau3AI is capable of cleaving the template despite of the hemimethylated state of the first CpG nucleotide. Similarly, HpaII can attack its unmethylated native site, leading to fragmentation of the template as long as no further methylation occurs. Impairment of Sau3AI cleavage indicates maintenance methylation activity, whereas a decrease in HpaII activity would result from <i>de novo</i> methylation. Therefore the assay is not only detecting activity of the enzyme but also includes its specificity for hemimethylated DNA over unmethylated DNA. |
| + | |||
| + | To quantify maintenance and de novo methylation activity, the 40-mer fragments remaining after DNMT1 incubation and restriction digest were separated by gel electrophoresis. The intensities of the detected bands corresponding to different fragments were measured using ImageJ Gel after background substraction. | ||
| Line 114: | Line 139: | ||
=Results= | =Results= | ||
| - | ==Optimizing expression and purification of linear mDNMT1(731-602)== | + | ==Optimizing expression and purification of linear mDNMT1 (731-602)== |
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=right| | align=right| | ||
caption=Figure 5) Optimisation of induction| | caption=Figure 5) Optimisation of induction| | ||
| - | descr=As soon as transformed Rosetta (DE3) cultures reached an OD600 of approximately 0.6, induction of linear mDNMT1(731-1602) was perfomed over night at either 15°C or 25°C, using different concentrations of IPTG. Higest expression of DNMT1 was observed at 15°C using 0.7mM IPTG.| | + | descr=As soon as transformed Rosetta (DE3) cultures reached an OD600 of approximately 0.6, induction of linear mDNMT1 (731-1602) was perfomed over night at either 15°C or 25°C, using different concentrations of IPTG. Higest expression of DNMT1 was observed at 15°C using 0.7mM IPTG.| |
file=DNMT1-Induction2.png}} | file=DNMT1-Induction2.png}} | ||
| - | Before starting circularization of DNMT1, we first tested if we can actually express and purify active linear DNMT1. Since DNMT1 expression in E.coli normally results in low yields (<1mg/liter of culture)[[#References|[15]]], we started with an optimization of the expression conditions. | + | Before starting circularization of DNMT1, we first tested if we can actually express and purify active linear DNMT1. Since DNMT1 expression in E.coli normally results in low yields (<1mg/liter of culture) [[#References|[15]]], we started with an optimization of the expression conditions. |
| - | In general, low temperatures and low IPTG concentrations are known to prevent the occurrence of insoluble inclusion bodies under conditions of overexpression. Thus, we tested different IPTG concentrations and temperatures during induction in order to maximize yields of active mDNMT1(731-1602). | + | In general, low temperatures and low IPTG concentrations are known to prevent the occurrence of insoluble inclusion bodies under conditions of overexpression. Thus, we tested different IPTG concentrations and temperatures during induction in order to maximize yields of active mDNMT1 (731-1602). We observed an optimal induction of our mDNMT1 construct after incubation at 15 °C using 0.7 mM IPTG (Figure 5). |
| - | + | After successful expression, the next critical step for recombinant protein purification is protein extraction. The lysis method has to be efficient but still gentle, especially when dealing with sensitive and low expressed proteins such as DNMT1. | |
| + | In order to optimize protein extraction we therefore compared cell lysis by sonification with disruption via French press (Figure 6A and 6B). Due to the low overall yield, mDNMT1 (721-1602) does not appear as a prominent band on the Coomassie-stained gel as normally expected for overexpressed recombinant proteins (Figure 6) . Since the efficiency of DNMT1 extraction could not be estimated from the Coomassie-stained gel, a western blot was performed. French press efficiently extracts soluble mDNMT1 (731-1602) whereas sonification leads to mDNMT1 accumulation in the pellet after ultracentrifugation (see Figure 6B, SN and pellet for sonification ). Indeed, the disruption of E.coli using French press has been reported to preserve the biological activity of susceptible proteins more efficiently than sonication [[#References|[18]]]. Therefore we used the French press for further extraction of linear as well as circular mDNMT1 (731-1602). | ||
| - | + | In the western blot, several other peptides with lower molecular weight were detected. These peptides possibly result from incomplete translation or degradation processes, since the construct used for initial mDNMT1 (731-1602) expression and purification (Figure 6) comprised an N-terminal hexa-histidine tag. To minimize the amount of incomplete protein products that would be enriched in the following purification, we optimized the DNMT1 construct by shifting the His-tag to the C-terminus. | |
| - | + | <div style="clear:both;"></div> | |
| - | + | ||
Further purification of the protein using a His-trap resulted in successful enrichment of mDNMT1f(731-1602) (Figure 6C). Corresponding eluates were pooled and further concentrated for use in functional assays. | Further purification of the protein using a His-trap resulted in successful enrichment of mDNMT1f(731-1602) (Figure 6C). Corresponding eluates were pooled and further concentrated for use in functional assays. | ||
| Line 138: | Line 163: | ||
align=left| | align=left| | ||
caption=Figure 6A) Comparison of DNMT1 extraction from bacteria by French press and sonification| | caption=Figure 6A) Comparison of DNMT1 extraction from bacteria by French press and sonification| | ||
| - | descr=mDNMT1(731-1602) expression in Rosetta (DE3) was performed over night at 15°C. Subsequently, the bacteria were lysed by sonification or French press and ultracentrifuged. After each step, samples were collected for analysis by SDS-PAGE. | + | descr=mDNMT1 (731-1602) expression in Rosetta (DE3) was performed over night at 15°C. Subsequently, the bacteria were lysed by sonification or French press and ultracentrifuged. After each step, samples were collected for analysis by SDS-PAGE. The resulting protein-membrane pellets and lysate supernatants were analyzed by SDS-PAGE. (Untransformed negative control (UnTra), intervals of cell disruption (1, 2), supernatant (SN)).| |
| - | (Untransformed negative control (UnTra), intervals of cell disruption (1, 2), supernatant (SN)).| | + | |
file=DNMT1-Celllysis.jpg}} | file=DNMT1-Celllysis.jpg}} | ||
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=right| | align=right| | ||
| - | caption=Figure 6B)Lysis via French press efficiently etracts mDNMT1(731-1602)| | + | caption=Figure 6B) Lysis via French press efficiently etracts mDNMT1 (731-1602)| |
| - | descr=Western Blot of | + | descr=Western Blot of lysates was performed with an anti-hexa-histidine antibody for specific detection of mDNMT1. Full length mDNMT1 (731-1602) is indicated with an arrow.| |
file=DNMT1-Celllysis-WB.jpg}} | file=DNMT1-Celllysis-WB.jpg}} | ||
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=left| | align=left| | ||
| - | caption=Figure 6C) Purification of linear mDNMT1(731-1602)| | + | caption=Figure 6C) Purification of linear mDNMT1 (731-1602)| |
| - | descr=Coomassie stained SDS-PAGE showing efficient purification of linear mDNMT1(731-1602) via His-column. Arrow indicates peptides corresponding to the molecular weight of mDNMT1(731-1602 | + | descr=Coomassie stained SDS-PAGE showing efficient purification of linear mDNMT1 (731-1602) via His-column. Arrow indicates peptides corresponding to the molecular weight of mDNMT1 (731-1602).| |
file=DNMT1-Purification-linear.png}} | file=DNMT1-Purification-linear.png}} | ||
<div style="clear:both;"></div> | <div style="clear:both;"></div> | ||
| - | ==Activity and specificity of linear mDNMT1(731-1602)== | + | ==Activity and specificity of linear mDNMT1 (731-1602)== |
| - | After successful expression and purification of linear DNMT1, we characterized the proteins activity and specificity for hemi-methylated DNA by performing the methylation activity assay described above. To test the maintenance methylation activity of our linear DNMT1, we performed the methylation assay with different incubation times of DNMT1. We observe an increasing amount of completely methylated template over time (Figure 7), represented by increasing amounts of template that are unsusceptible towards SauAI cleavage. | + | After successful expression and purification of linear DNMT1, we characterized the proteins activity and specificity for hemi-methylated DNA by performing the methylation activity assay described above. To test the maintenance methylation activity of our linear DNMT1, we performed the methylation assay with different incubation times of DNMT1 (731-1602). We observe an increasing amount of completely methylated template over time (Figure 7), represented by increasing amounts of template that are unsusceptible towards SauAI cleavage. |
| - | The truncated DNMT1 (731-1602) showed maximal methylation of the template after approximately | + | |
| + | The truncated DNMT1 (731-1602) showed maximal methylation of the template after approximately 30min. Therefore, our data agrees with efficiencies that have been reported in literature for <i>wild type</i> and truncated versions of mDNMT1 [[#References|[15]]][[#References|[16]]] .Overall, the reaction kinetics of the linear protein seem to be promising for later use in a '''"PCR2.0"''', assuming that circularization of the protein will not ablate its function. | ||
| + | |||
| + | In oder to be used in the targeted '''"PCR2.0"''' mDNMT1 needs to be highly specific for hemi-methylated sites and should not display <i>de novo</i> methylation activity. Therefore, we included HpaII digestion in our assay to determine <i>de novo</i> methylation of CpG sites. The first results prove that the purified mDNMT1 (731-1602) does not exhibit <i>de novo</i> methylation activity (Figure 8). Even for increasing incubation times, HpaII is not repressed which indicates absence of <i>de novo</i> methylation. | ||
| + | |||
| + | However, the analysis of the gel pictures is difficult since restriction efficiency of HpaII was not 100% as it can be seen from the hemimethylated control. Therefore, uncut template is possibly not resulting from unspecific methylation activity of the enzyme. Further tests, especially with the circularized protein will have to be performed to confirm these initial data. | ||
| + | <div style="clear:both;"></div> | ||
| + | |||
| + | <div style="clear:both;"></div> | ||
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=left| | align=left| | ||
| - | caption=Figure | + | |
| - | descr= Purified linear mDNMT1(731-1602) was incubated with 40mer DNA template (see Figure4) for indicated times at 37°C. Subsequently the samples were digested with Sau3AI. Fragments were separated by PAGE and stained with ethidium bromide. (Homo me= homomethylated substrate, control for enzyme inhibition through methylation, Hemi me=hemimethylated substrate positive control for digest).| | + | |
| + | caption=Figure 7A) Maintenance Methylation activity of linear Dnmt1 (731-1602) over time| | ||
| + | descr= Purified linear mDNMT1 (731-1602) was incubated with 40mer DNA template (see Figure4) for indicated times at 37°C. Subsequently the samples were digested with Sau3AI. Fragments were separated by PAGE and stained with ethidium bromide. (Homo me= homomethylated substrate, control for enzyme inhibition through methylation, Hemi me=hemimethylated substrate positive control for digest).| | ||
file=activityovertime.jpg}} | file=activityovertime.jpg}} | ||
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
| - | align= | + | align=left| |
| - | caption=Figure | + | caption=Figure 7B) Quantification of maintenance methylation activity of linear Dnmt1 (731-1602) over time| |
| - | descr=Purified linear mDNMT1(731-1602) was incubated with 40mer DNA template (see Figure4) for indicated times at 37°C. Subsequently the samples were digested with HpaII. Fragments were separated using gel-electrophoresis and visualized with ethidiumbromid staining. (Homo me= homomethylated substrate,control for enzyme inhibition through methylation, Hemi me=hemimethylated substrate positive control for digest).| | + | descr= Amount of methylation was calculated by normalizing the intensity of the upper band to the overall intensity of both bands. Graph shows technical duplicate of the assay. All measurements were performed with ImageJ.| |
| + | file=dnmt1_act_maint_over_time.png}} | ||
| + | <div style="clear:both;"></div> | ||
| + | {{:Team:Heidelberg/templates/image-half| | ||
| + | align=left| | ||
| + | |||
| + | |||
| + | caption=Figure 8A) <i>De novo</i> methylation activity of mDNMT1| | ||
| + | descr=Purified linear mDNMT1 (731-1602) was incubated with 40mer DNA template (see Figure4) for indicated times at 37°C. Subsequently the samples were digested with HpaII. Fragments were separated using gel-electrophoresis and visualized with ethidiumbromid staining. (Homo me= homomethylated substrate,control for enzyme inhibition through methylation, Hemi me=hemimethylated substrate positive control for digest).| | ||
file=specificityovertime.jpg}} | file=specificityovertime.jpg}} | ||
| + | {{:Team:Heidelberg/templates/image-half| | ||
| + | align=right| | ||
| + | caption=Figure 8B) Quantification of linear mDNMT1 <i>de novo</i> methylation activity| | ||
| + | descr=Amount of methylation was calculated by normalizing the intensity of the upper band to the overall intensity of both bands. All measurements were performed with ImageJ.| | ||
| + | file=dnmt1_act_de_novo_over_time.png}} | ||
| - | |||
| - | |||
<div style="clear:both;"></div> | <div style="clear:both;"></div> | ||
| - | =Circular mDNMT1(731-1602) - The pivotal element of PCR2.0= | + | =Circular mDNMT1 (731-1602) - The pivotal element of PCR2.0= |
| - | ==Expression and purification of mDNMT1(731-1602) for circularization== | + | ==Expression and purification of mDNMT1 (731-1602) for circularization== |
After we succeeded in purifying active and specific linear mDNMT1, we went on with cloning, expression and purification of the circular DNMTs. | After we succeeded in purifying active and specific linear mDNMT1, we went on with cloning, expression and purification of the circular DNMTs. | ||
| - | Expression and purification of mDNMT1(731-1602) for the intein as well as the sortase approach were conducted as they had been established for the linear DNMT1. Elutions from the His-Trap showed an enrichment and concentration of peptides that correlate with the expected molecular weight of the truncated mDNMT1(731-1602) (Figure 9A and 9B). Nevertheless, purification with a His-Trap alone does not seem to exclude a variety of impurities that either result from degraded mDNMT1 that still contains a His-Tag or unspecific binding to the affinity column. Advanced protocols as they were used by Song and collegues [[#References|[19]]] for analysis of the proteins crystal structure are therefore including several more steps. Nevertheless, additional steps of purification increase the risk of reducing the overall protein yield and activity, necessitating greater amounts of starting material. Since the active protein should already be included and efficiently enriched in our sample despite of present impurities, we continued with the functional analysis of the sample. | + | Expression and purification of mDNMT1 (731-1602) for the intein as well as the sortase approach were conducted as they had been established for the linear DNMT1. Elutions from the His-Trap showed an enrichment and concentration of peptides that correlate with the expected molecular weight of the truncated mDNMT1 (731-1602) (Figure 9A and 9B). Nevertheless, purification with a His-Trap alone does not seem to exclude a variety of impurities that either result from degraded mDNMT1 that still contains a His-Tag or unspecific binding to the affinity column. Advanced protocols as they were used by Song and collegues [[#References|[19]]] for analysis of the proteins crystal structure are therefore including several more steps. Nevertheless, additional steps of purification increase the risk of reducing the overall protein yield and activity, necessitating greater amounts of starting material. Since the active protein should already be included and efficiently enriched in our sample despite of present impurities, we continued with the functional analysis of the sample. |
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=right| | align=right| | ||
| - | caption=Figure 9A) Efficiency of mDNMT1(731-1602) purification for the intein approach| | + | caption=Figure 9A) Efficiency of mDNMT1 (731-1602) purification for the intein approach| |
| - | descr=Induction of Rosetta (DE3) and purification were performed as described above. After each step, samples were collected for analysis by Coomassie stained SDS-PAGE. Samples are numbered according to their chronology of collection. Arrow indicates mDNMT1(731-1602).| | + | descr=Induction of Rosetta (DE3) and purification were performed as described above. After each step, samples were collected for analysis by Coomassie-stained SDS-PAGE. Samples are numbered according to their chronology of collection. Arrow indicates mDNMT1 (731-1602).| |
file=DNMT1-intein-Purification3.png}} | file=DNMT1-intein-Purification3.png}} | ||
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=right| | align=right| | ||
caption=Figure 9B) Sortase approach - Efficiency of Purification| | caption=Figure 9B) Sortase approach - Efficiency of Purification| | ||
| - | descr=Induction of Rosetta (DE3) and purification were performed as described above. After each step, samples were collected for analysis by Coomassie stained SDS-PAGE. Samples are numbered according to their chronology of collection. Arrow indicates mDNMT1(731-1602).| | + | descr=Induction of Rosetta (DE3) and purification were performed as described above. After each step, samples were collected for analysis by Coomassie stained SDS-PAGE. Samples are numbered according to their chronology of collection. Arrow indicates mDNMT1 (731-1602).| |
file=DNMT1-sortase-Purification2.png}} | file=DNMT1-sortase-Purification2.png}} | ||
| Line 196: | Line 241: | ||
align=right| | align=right| | ||
caption=Figure 10) Expression and Purification of intein and sortase constructs with different linkers| | caption=Figure 10) Expression and Purification of intein and sortase constructs with different linkers| | ||
| - | descr=Expression and Purification of different constructs in Rosetta (DE3) was performed as described above. Unpurified as well as purified samples were collected for analysis by Coomassie stained SDS-PAGE. Ctrl indicates the untransformed control. D= no linker, R= Rigid linker, F=Flexible linker, L= linear. Arrow indicates peptides with molecular weight corresponding to mDNMT1(731-1602). Asterix indicates possibly spliced Intein constructs.| | + | descr=Expression and Purification of different constructs in Rosetta (DE3) was performed as described above. Unpurified as well as purified samples were collected for analysis by Coomassie-stained SDS-PAGE.(Ctrl indicates the untransformed control. D= no linker, R= Rigid linker, F=Flexible linker, L= linear DNMT1. Arrow indicates peptides with molecular weight corresponding to mDNMT1 (731-1602). Asterix indicates possibly spliced Intein constructs).| |
file=DNMT1-Purification1.png}} | file=DNMT1-Purification1.png}} | ||
| - | Our approach of heat-stabilization includes the testing of different linkers that had been calculated by our [https://2014.igem.org/Team:Heidelberg/Software/Linker_Software CRAUT Linker Software]. We tested a flexible linker, consisting mostly of glycines and serines and a rigid linker, which was introduced to maximize stabilization of the protein through cicularization. Purification of the mDNMT1(731-1602) variants with different linkers for circularization was successful (Figure 10), but the yields were lower compared to different batches of linear mDNMT1(731-1602) that had been produced in a similar way. | + | Our approach of heat-stabilization includes the testing of different linkers that had been calculated by our [https://2014.igem.org/Team:Heidelberg/Software/Linker_Software CRAUT Linker Software]. We tested a flexible linker, consisting mostly of glycines and serines and a rigid linker, which was introduced to maximize stabilization of the protein through cicularization. Purification of the mDNMT1 (731-1602) variants with different linkers for circularization was successful (Figure 10), but the yields were lower compared to different batches of linear mDNMT1 (731-1602) that had been produced in a similar way. |
| - | + | ||
| - | + | Interestingly, a shift between mDNMT1 (731-1602) expressed from the intein construct compared to the sortase or linear versions was detected. Since protein circularization via inteins is an autocatalytic process that takes place inside the bacteria, this shift could possibly result from successful splicing activity of the inteins. The resulting product lacks Smt3 and is therefore approximately 30kDa smaller than its precursor (Figure 10, indicated by an asterix). Since intein splicing likely occurred, flexible- and rigid-linked-DNMT1 could be circularized. | |
| - | + | To prove circularization by inteins, we cooperated with the core facility "Protein Analysis" of the German Cancer Research Center (DKFZ). In the facility, circularized and linear DNMT1 were extracted from a provided gel, digested with trypsin and analyzed by ESI mass spectrometry. Unfortunately, we do not have the final results yet but are confident to be able to present them at the jamboree. | |
| + | |||
| + | In contrast to the intein DNMT1 construct, mDNMT1 (731-1602) with sortase recognition sequences needs to be further processed before a circular protein is obtained. Unfortunately, TEV cleavage of the purified protein, which is necessary for subsequent circularization with sortase was not very efficient. Therefore sortase treatment did not yield in a detectable amount of processed mDNMT1 (731-1602). Therefore, we fully concentrated on the promising intein approach for further mDNMT1 (731-1602) circularization. | ||
<div style="clear:both;"></div> | <div style="clear:both;"></div> | ||
| - | ==Thermal stability of circular | + | ==Thermal stability of circular mDNMT1s(731-1602)== |
| + | |||
| + | In our final assay we compared the effect of a PCR-relevant heat shock on the activity of linear and circular DNMT1s. Thereby, we wanted to examine the ability of circular DNMT1 to withstand higher temperatures and the feasibility of a PCR 2.0 using circularized DNMT1. Linear DNMT1 (L) as well as rigid linker (RC) and flexible linker-containing (FC) DNMT1s were exposed for 5s to temperatures between 55°C and 72°C that would also be reached in a PCR and subsequently analyzed for methylation activity. | ||
| + | |||
| + | In the gel picture (Figure 11A and B) a darker upper band in the FC sample compared to the linear DNMT1 sample indicates higher methylation activity of circular DNMT1 with flexible Linker at all temperatures Quantification of the band intensities using Image J shows that activity of circular DNMT1 with the flexible linker (black) is exceeding the linear and circular with rigid linker at least by a factor of 2. Surprisingly, the methylation activity of circular DNMT1 with a flexible linker seems to increase with higher temperatures. | ||
| + | |||
| + | For significant evaluation, more data has to be collected. When applying Michael Menten kinetics, the methylation reaction for the controls already approaches saturation. Therefore, the normalization to the original activity might be misleading. For further improvement of the assay, less volume of DNMT1 has to be taken for the enzyme reaction. | ||
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=right| | align=right| | ||
| - | caption=Figure | + | caption=Figure 11A) Higher maintenance methylation of circular mDNMT1 (731-1602) with flexible linker after heat-shock| |
descr=Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 55°C and 60°C. | | descr=Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 55°C and 60°C. | | ||
file=heatshock55-60.jpg}} | file=heatshock55-60.jpg}} | ||
{{:Team:Heidelberg/templates/image-half| | {{:Team:Heidelberg/templates/image-half| | ||
align=right| | align=right| | ||
| - | caption=Figure | + | caption=Figure 11B) Higher maintenance methylation activity of ciruclar mDNMT1 with flexible linker after heat-shock at PCR-relevant temperatures.| |
| - | descr=Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 65°C and 72°C.Methylation after one hour was measured. | + | descr=Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 65°C and 72°C.Methylation after one hour was measured. The dark upper band for FC indicates higher methylation activity.| |
file=heatshock65-72.jpg}} | file=heatshock65-72.jpg}} | ||
| + | |||
| + | <div> | ||
| + | {{:Team:Heidelberg/templates/image-full| | ||
| + | align=right| | ||
| + | caption=Figure 11C) Amount of methylation activity kept after heat shock | | ||
| + | descr=Circular DNMT1 with rigid linker (RC) in blue, circular DNMT1 with flexible linker (FC) in red and linear DNMT1 (L) in blue over a temperature range from 55°C to 72°C.| | ||
| + | file=dnmt1_heatshock.png}} | ||
| + | </div> | ||
| + | |||
| + | ==Outlook== | ||
| + | |||
| + | Our next steps until the jamboree will be to prove efficient linkage of N- and C- terminus for our circularized mDNMT1 (731-1602) via mass spectrometry. Further methylation activity assays with circularized protein are planned to validate significance of the obtained data. Moreover, we are looking forward to use our heat-stable mDNMT1 (731-1602) in the first '''”PCR2.0”'''. | ||
=Discussion= | =Discussion= | ||
| + | |||
| + | == Achievements and impact== | ||
| + | Taken together, we could show that intein-mediated circularization of mDNMT1 (731-1602) conceivably increased heat stability of the enzyme without impeding specific methylation activity. We proved that circularization with inteins is an easy method to increase heat stability of complex and sensitive proteins like mDNMT1 (731-1602). In comparison, other methods used for protein stabilization such as introduction of disulfide bonds or hydrophobic amino acids, require sophisticated structure analysis or non-physiological reaction conditions [[#References|[20]]][[#References|[21]]]. In fact, mDNMT1 (731-1602) is the largest protein that has ever been circularized so far and even more might follow. | ||
| + | |||
| + | Our [https://2014.igem.org/Team:Heidelberg/Software/Linker_Software CRAUT Linker Software] was essential to achieve circularization of mDNMT1 (731-1602) while preserving its natural function. In first experiments, the two designed linker compositions resulted in divergent heat stabilities of the protein. Our software successfully narrowed down the infinite amount of possibilities to circularize mDNMT1 (731-1602). Further experimental data of enzyme-specific linker variants will help to further improve the software predictions. Circularized mDNMT1 (731-1602) represents a valuable tool for the realization of a methylation maintaining '''”PCR2.0”'''. This foundational advance aims to enable broad functional analysis of DNA methylation patterns without prior knowledge on methylation frequency or dependency on other methods. For the detection of DNA methylation using bisulfite sequencing, handling of limited amounts of primary material still represents a major challenge: | ||
| + | |||
| + | {| class="table table-hover" | ||
| + | |- | ||
| + | | || ||| |||| ''“Future developments will undoubtedly allow information to be recovered from most genomic CpGs, the key being the ability to amplify DNA before bisulfite conversion. The capacity to capture the DNA methylome from individual cells will be critical for a full understandingof early embryonic development, cancer progression and generation of induced pluripotent stem cells.”'' - Smallwood et al., ''Nat. Methods (20014)'' [[#References|[29]]]||||| |||||| | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | Approaches including single molecule sequencing have been reported, but have not been established in an affordable and efficient manner yet [[#References|[27]]]. Furthermore, these methods are limited to the detection of methylation patterns and do not allow functional analysis without complete analysis and cost-intensive synthesis. Therefore, our '''”PCR2.0”''' aims to revolutionize the field of epigenetics by linking complex methylation patterns to their actual function. <i>In vitro</i> as well as <i>in vivo</i> experiments, requiring large amounts of methylated DNA will be feasible by using our '''”PCR2.0”'''. Those include for example the detection of DNA methylation binding proteins using Chromatin Immunoprecipitation or Electrophoretic Mobility Shift Assays. Further functional studies could investigate cellular regulation upon transfection with methylated template. | ||
| + | |||
| + | A very interesting application for the pharmaceutical use of our '''"PCR2.0"''' could be the large scale production of methylated gene therapy vectors. Since methylation of gene transfer vectors has been reported to extend transgene product expression by lowering cellular immunogenicity, the efficiency gene therapies might be improved with our method.[[#References|[22]]] | ||
| + | |||
| + | ==Considerations for further PCR2.0 development== | ||
| + | Since significance of the obtained methylation activity data still has to be proven in biological replicates, we could not yet precisely quantify the effect of DNMT1 stabilization by circularization. Therefore further steps in addition to the circularization of mDNMT1 (731-1602) could be necessary to obtain the required heat stability and specificity for our methylation maintaining '''"PCR 2.0"'''. Those for example include further protein stabilization by enhancing the hydrophobicity of the protein core or by introducing direct links to limit possible conformations [[#References|[23]]]. | ||
| + | |||
| + | Nevertheless, it is unlikely that mDNMT1 (731-1602) can be stabilized in a way to bear temperatures of 95°C, which are essential for conventional PCR. Therefore, another upcoming topic is the method of low temperature PCRs that exploit the function of helicases to segregate DNA strands. Even though these PCRs can be performed at lower temperatures than conventional PCRs, none of them has reached widespread establishment yet. Main reasons are that isothermal PCRs require either too long incubation times per cycle [[#References|[24]]] or still exceed physiological temperatures of most enzymes, introducing new candidates for circularization... [[#References|[25]]] | ||
| + | |||
| + | Despite enduring activity of mDNMT1 (731-1602) at high temperatures, the heat-stability of its cofactors also plays a crucial role for establishing a '''”PCR 2.0”'''. Here, a still challenging factor is methyl-donor S-adenosyl methionine (SAM) that exhibits only low stability at high temperatures [[#References|[30]]]. Nevertheless, addition of the quite cheap methyl-donor after each cycle of replication could circumvent this problem until a more stable variant of the cofactor is available. | ||
| + | |||
| + | If there are occurring problems with the specificity of the enzyme at higher temperatures which have not been assessable from our data, the '''“PCR 2.0”''' could be complemented with enzymes such as UHRF1, that have been reported to increase the specificity of mDNMT1 (731-1602) [[#References|[31]]]. Wild type DNMT1 is known to be associated to the replication complex for direction of its activity. Design of an appropriate Polymerase-DNMT-fusion protein could imitate this connection and therefore further reduce stochastic methylation events [[#References|[26]]]. | ||
| + | |||
| + | Overall, the iGEM Team Heidelberg has achieved important milestones in establishing a methylation maintaining '''"PCR 2.0"''' by expressing and purifying the largest circular protein ever. Despite of its use in a '''"PCR 2.0"''', the stabilized mDNMT1 (731-1602) can also be used in other methylation assays. | ||
=References= | =References= | ||
| - | [1] | + | [1] Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–70 (2008). |
| - | [2] | + | [2] Watanabe, M., Yuzawa, H., Handa, N., & Kobayashi, I.. Hyperthermophilic DNA methyltransferase M.PabI from the archaeon Pyrococcus abyssi. Applied and Environmental Microbiology, 72(8), 5367–75 (2006). |
[3] Vieille, C. & Zeikus, G. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65, 1–43 (2001). | [3] Vieille, C. & Zeikus, G. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65, 1–43 (2001). | ||
| Line 261: | Line 351: | ||
[19] Song, J., Rechkoblit, O., Bestor, T. H. & Patel, D. J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–40 (2011). | [19] Song, J., Rechkoblit, O., Bestor, T. H. & Patel, D. J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–40 (2011). | ||
| + | |||
| + | [20] Betz, S. F. Disulfide bonds and the stability of globular proteins. 1551–1558 (1993). | ||
| + | |||
| + | [21] Bulaj, G. Formation of disulfide bonds in proteins and peptides. Biotechnol. Adv. 23, 87–92 (2005). | ||
| + | |||
| + | [22] Reyes-Sandoval, a & Ertl, H. C. J. CpG methylation of a plasmid vector results in extended transgene product expression by circumventing induction of immune responses. Mol. Ther. 9, 249–61 (2004). | ||
| + | |||
| + | [23] Vieille, C. & Zeikus, G. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65, 1–43 (2001). | ||
| + | |||
| + | [24] Vincent, M., Xu, Y. & Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 5, 795–800 (2004). | ||
| + | |||
| + | [25] Xu, G. et al. Cross priming amplification: mechanism and optimization for isothermal DNA amplification. Sci. Rep. 2, 246 (2012). | ||
| + | |||
| + | [26] Vilkaitis, G., Suetake, I., Klimasauskas, S. & Tajima, S. Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J. Biol. Chem. 280, 64–72 (2005). | ||
| + | |||
| + | [27] Krueger, F., Kreck, B., Franke, A. & Andrews, S. R. DNA methylome analysis using short bisulfite sequencing data. Nat. Methods 9, 145–51 (2012). | ||
| + | |||
| + | [28] Smith, Z. D. & Meissner, A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–20 (2013). | ||
| + | |||
| + | [29] Smallwood, S. a et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 6–11 (2014). | ||
| + | |||
| + | [30] Matos, J. & Wong, C. S-adenosylmethionine: Stability and stabilization. Bioorg. Chem. 80, 71–80 (1987). | ||
| + | |||
| + | [31] Bashtrykov, P., Jankevicius, G., Jurkowska, R. Z., Ragozin, S. & Jeltsch, A. The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J. Biol. Chem. 289, 4106–15 (2014). | ||
Latest revision as of 03:39, 18 October 2014

Abstract
DNA methylation is the most abundant DNA modification and essential for embryonic development, gene regulation and genomic stability. Although several methods for the detection of methylation patterns exist, there is no easy way to amplify methylated DNA for in vitro or in vivo studies.
To empower epigenetic research, we envisioned a PCR2.0 which maintains DNA methylation patterns during amplification. The central element of this PCR2.0 is a heat-resistant DNA methyltransferase - DNMT1 (731-1602) - which we created by circularization using our intein toolbox and the CRAUT linker software.
So far, our DNMT1 represents the largest circularized protein which highlights the usefulness of our intein toolbox in combination with the CRAUT linker software. Increased heat resistance of our circular DNMT1 which was observed in initial assays smoothens the path for the establishment of a PCR2.0 and illustrates the suitability of intein-mediated circularization for the advancement of heat resistant proteins.
Highlights
- circularization seems to increase heat stability of mDNMT1 (731-1602): successful first step towards PCR2.0.
- mDNMT1 is the largest circularized protein (105kDa) so far.
- efficient expression and purification of active linear and circular mDNMT1 (731-1602) in E.coli.
- successful application of CRAUT linker design software and intein toolbox for circularization.
Contents |
Introduction
Motivation
The invention of the polymerase chain reaction in 1983 by Kary Mullis revolutionized our world by enabling amplification of DNA in an exponential manner. Further improvements including the use of thermo-stable DNA polymerases made the method even more efficient, allowing its widespread use in nearly every field of modern diagnostics and research. However, a major part of information is lost when using conventional PCR as DNA modifications are not copied. The most abundant modification is DNA methylation, which is a prominent regulator of gene expression in all kingdoms of life. Methylation and other modifications that influence the DNA function without changing its actual sequence are studied in the fast-growing field of epigenetics. Even though the detection and mapping of methylation patterns have become feasible in a high-throughput manner by using bisulfite sequencing and array techniques [1], further functional analysis is still hindered by the small amount of primary material. Up to now, there is no method available to amplify methylated DNA without knowledge of methylation patterns and expensive de-novo synthesis.
To empower epigenetics, we propose the “PCR 2.0” as an easy and efficient way to amplify DNA templates in an exponential manner while maintaining their specific methylation pattern. The pivotal element of this PCR reaction is a heat-stable DNA methyltransferase (DNMT). Although comparable enzymes exist in thermophile organisms [2], until now no suitable protein has been found or synthesized that withstands the harsh conditions of a PCR.
To achieve this goal of a PCR2.0 that can be used to amplify DNA including its intrinsic methylation pattern, our main aim was the generation of a heat-stable Dnmt1 that can be produced in a larger scale and shows efficient and specific methylation of DNA. It was reported that the insertion of additional intramolecular bonds can increase the stability of proteins [3][4], and that specifically joining the C- and N-terminus improves the thermostability of smaller peptides [5]. This is why our approach to generate a heat-stable Dnmt1 applies:
- protein circularization using our intein toolbox
- creating a potent Software to design efficient linker peptides connecting C- and N-terminus
To enable the PCR2.0 we started the circularization of the largest protein so far…
Epigenetics - there is more than just A, T, C and G…
In mammals and other vertebrates DNA methylation occurs at the C5 position of cytosine nucleotides that are followed by guanines (CpG). It is a prominent key regulator of transcription, embryonic development, X chromosome inactivation and many other cellular functions [6][7]. The human genome contains about 28 million CpGs of which 60-80% are methylated [28]. Due to its great prevalence and the heredity of this modification, the 5-methylcytosine (5mC) is also known as the “fifth base” of eukaryotic genomes [9]. DNA methylation is preferentially occurring at intergenic regions and repetitive sequences, where it is known to silence gene expression [10][11][12]. Inappropriate cytosine methylation and silencing has been reported to play a crucial role in the development of numerous diseases, including cancer, imprinting diseases and repeat-instability based diseases such Huntington's disease [13].
A closer look at DNA Methyltransferases
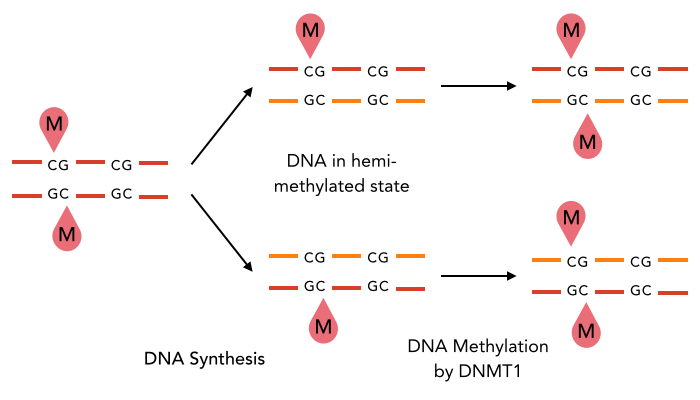
The enzyme DNMT1 is of particular interest for methylation maintenance PCR, as it allows exact duplication of the original methylation pattern to every new DNA copy.
The family of enzymes called DNA methyltransferases (DNMTs) is responsible for the establishment and maintenance of cell-type specific DNA methylation patterns. Whereas the two enzymes DNMT3a and DNMT3b contribute to de novo methylation of DNA during development, DNMT1 is preserving existing methylation patterns throughout cell divisions. To do so, DNMT1 exploits the principle of semi-conservative replication, using the parental strand as a template to create an exact copy of methylation patterns on the daughter strand (Figure 1). After DNA synthesis, the DNA consists of a methylated and an unmethylated strand. DNMT1 recognizes these so called hemi-methylated CpG sites, and transfers a methyl group from the methyl donor S-Adenosyl-methionine (SAM) to the cytosine belonging to the newly synthesized strand [15][19].

Structural overview of full length mDNMT1 and truncated mDNMT1 (731–1602) (A) Color-coded schematic overview of domain structure and numbering of mDNMT1 sequence. (B) Ribbon presentation of full-length DNMT1 with the CXXC, BAH1, BAH2,and methyltransferase domain colored in light blue, yellow, orange and red, respectively. (C) Crystal structure of truncated DNMT1, missing the CXXC domain and schowing more adjacent protein termini that the full-length version.
The complex structure of DNMT1 and several truncated versions have been recently solved by X-ray crystallography [19]. For our experiments we used the smallest truncated version of DNMT1 - mDNMT1 (731-1602) - that has been reported to efficiently mediate methylation maintenance. This truncated version is derived from Mus musculus and comprises the amino acids 731-1602 of the full length protein [15][19] (kindly provided by Dr. Bashtrykov after approval from Prof. Patel). Murine DNMT1 (731-1602) contains a C-terminal catalytic methyltransferase domain as well as the bromo-adjacent homology (BAH) domains, which prevent the binding of unmethylated DNA to the catalytic core. The truncated enzyme is missing the CXXC domain, which has a high affinity to hemi-methylated CpG dinucleotides, but was shown to be dispensable for protein function [16]. In contrast to the full-length DNMT1, mDNMT1 (731-1602) has the great advantage of being efficiently expressed in E.coli. Another benefit of the truncated DNMT1 is that the N- and C- terminus are closer together than in the full-length DNMT1 (Figure 2). Thus circularization might cause less deformation and show lower impact on the overall activity, which makes this truncated mDNMT1 an ideal candidate for our purposes.
Circularization - The missing link
Heat stabilization of mDNMT1 as a pivotal element of our methylation-maintaining PCR2.0 was approached by circularization of the protein. Until now, only smaller proteins have been stabilized using this method. Therefore, to our knowledge, mDNMT1 – even in its shortest truncated version of 871 amino acids and a molecular weight of approximately 100kDa – is the largest protein that has ever been tried to circularize. According to the crystal structure, the distance between the termini of the truncated DNMT1 is 48 Å. We suspected that circularization of the protein by direct fusion of both termini might cause deformation of the protein structure. Therefore we used our own CRAUT Linker Software to create a linking amino acid sequence, adapted to the structure of DNMT1. The software is able to design protein linkers with the required length to bridge the gap between the protein termini while bypassing the catalytic core of the enzyme.
Since we could not estimate the impact of the linker introduction, we focused on two different kinds of linkers: a so called rigid linker that has been calculated and optimized during the establishment of our linker software and a flexible peptide connection consisting mostly of glycine and serine. To perform circularization we have used the split NpuDnaE Intein, since it is used as the golden standard for protein splicing and has been efficiently tested by us through circularization of GFP, Lysozyme and Xylanase. Moreover, we tried to implement protein circularization by using sortase A, which is recognizing and cleaving a carboxyterminal sorting signal followed by a transpeptidation reaction that can be exploited for protein circularization. This method has been reported to be very efficient [4] and was therefore included in our study as possible candidate for large scale productions of circular proteins.
Experimental procedures
Constructs
We designed constructs in order to characterize the efficiency of sortase A as well as intein mediated circularization of DNMT1. Both approaches are based on the comparison of the circularized protein with a corresponding linear counterpart at different temperatures and incubation times.
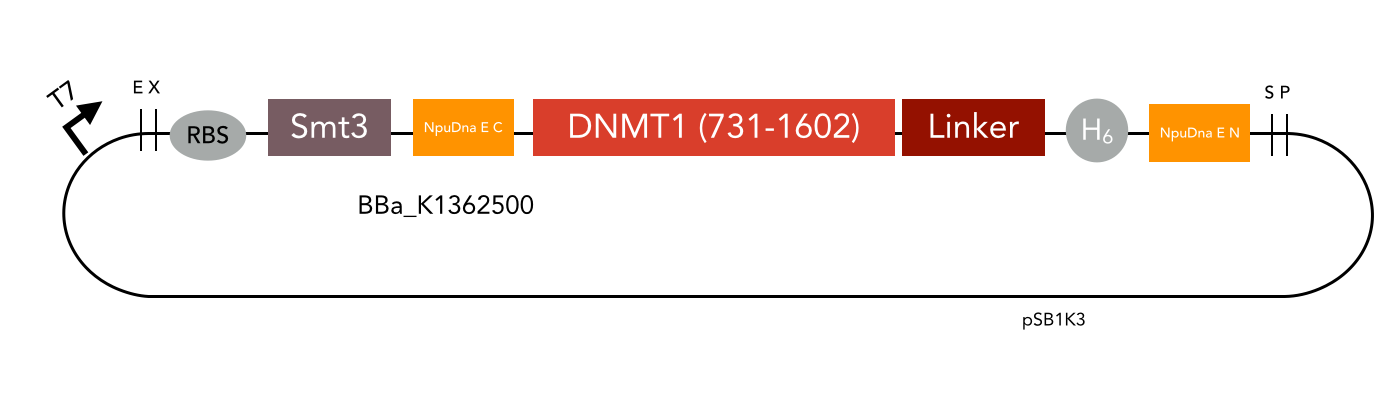
The mDNMT1 (731-1602) constructs that have been designed to allow sortase A mediated cirularization are flanked by the sortase A recognition sequence LPETGG on the C–terminus as well as N-terminal glycines that that can be exposed through previous TEV treatment. Since the transpeptidation via sortase requires an additional in vitro reaction, subsequent purification is necessary. To facilitate this step and to enrich the circular product in the sample, a hexa-histidine tag is present in the initial protein that is lost during circularization. Hence, an additional affinity chromatography via His trap can be used to separate the histidine tag containing sortase and not circularized educt from the circular flow-through.
All cloning steps were carried out using our sortase standard.
The following linkers that had been optimized along with our CRAUT Linker Software:
| Definition | Sequence |
|---|---|
| Rigid linker | GGAEAAAKEAAAKVNLTAAEAAAKEAAAKEAAAKEAAAKEAAAKAVNLTAAEAAAKAHHHHHHSGRGT |
| Flexible linker | CWEGGGSGGGSGGGSGGGSGGGSGGGSGGGSGGGSGGGSGGGSHHHHHHSGRGT |
Expression and purification
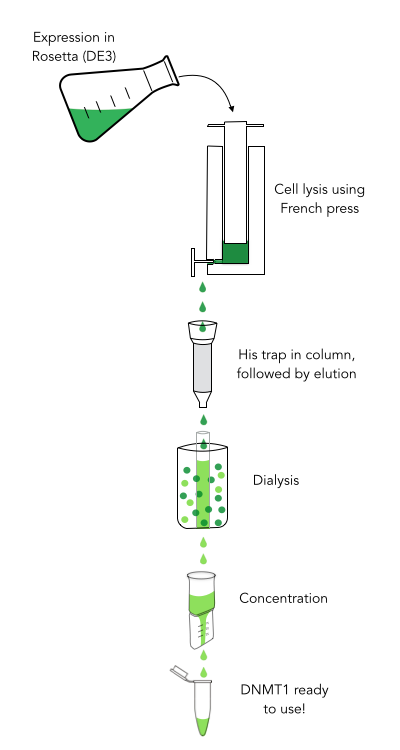
The purification of recombinantly expressed mDNMT1 is a challenging task including large scale induction, efficient cell lysis, reduction of sample volume via ultracentrifugation, immobilized Ni ion affinity chromatography, dialysis and concentration through centrifugation.
In comparison to the full length murine DNMT1, our modified mDNMT1 (731-1602) can be expressed in E.coli (Rosetta DE3) and does not need the establishment of Baculovirus based system and the use of insect cell cultures. First experiments were conducted to determine the optimal conditions for protein induction and expression. We tested several concentrations of IPTG as well as different temperatures to maximize the yield of mDNMT1. Still, even though expression of the constructs can be simplified by using E.coli, purification of the recombinant protein remains a challenging task requiring multiple different steps that had to be adapted and established from published protocols [19].
In a first step, we optimized the process of cell lysis, to be able to extract a maximum amount of active protein from our cultures. Therefore we compared the two commonly used methods sonication and disruption via French press. Subsequently, the sample was cleared from cell debris by ultracentrifugation. The protein of interest containing a hexa-histidine tag was further enriched via immobilized metal ion affinity chromatography (IMAC) using a His trap and eluted with imidazol. In a next step, low molecular weight solutes such as salts and imidazol that would interfere with protein function were removed from the sample by dialysis. Finally, the purified protein was concentrated by size-exclusion centrifugation using filters with an appropriate mass cutoff.
Methylation activity assay

Sequence of the 40-mer DNA template. CpG dinucleotides are shaded in gray, Sau3AI and HpaII recognition sites are depicted in red. The first CpG dinucleotide is methylated on the reverse strand.
To measure the efficiency and specificity of mDNMT1 maintenance methylation activity, we used an assay that is based on the two methylation-sensitive restriction enzymes Sau3AI and HpaII. It relies on the inhibition of restriction enzymes when methyl groups are attached to the cleavage site. We used a 40-mer DNA template with one hemi-methylated and one unmethylated CpG site which are located within the Sau3AI and HpaII restriction site, respectively (adapted from Bashtrykov et colleagues [16].
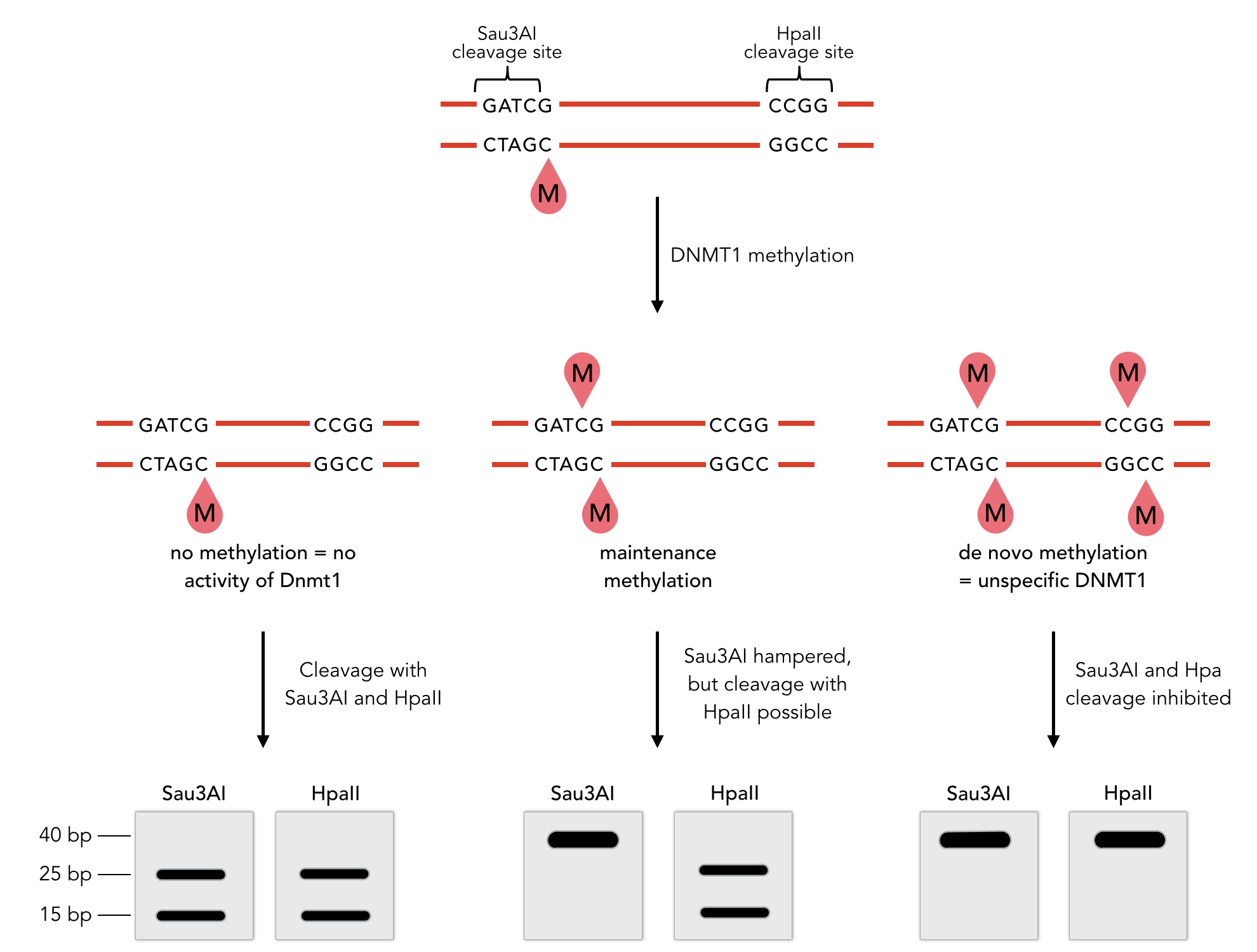
Principle of mDNMT1 methylation assay using a 40-mer substrate with Sau3AI and HpaII cleavage sites that are blocked upon further CpG methylation. The lack of methylation activity leads to fragmentation of the DNA template with either of the two restriction enzymes. Whereas specific methylation of hemimethylated CpGs hampers only cleavage by Sau3AI, de novo methylation activity of mDNMT1 can be detected when HpaII cleavage is blocked as well.
The restriction enzyme Sau3AI is capable of cleaving the template despite of the hemimethylated state of the first CpG nucleotide. Similarly, HpaII can attack its unmethylated native site, leading to fragmentation of the template as long as no further methylation occurs. Impairment of Sau3AI cleavage indicates maintenance methylation activity, whereas a decrease in HpaII activity would result from de novo methylation. Therefore the assay is not only detecting activity of the enzyme but also includes its specificity for hemimethylated DNA over unmethylated DNA.
To quantify maintenance and de novo methylation activity, the 40-mer fragments remaining after DNMT1 incubation and restriction digest were separated by gel electrophoresis. The intensities of the detected bands corresponding to different fragments were measured using ImageJ Gel after background substraction.
Results
Optimizing expression and purification of linear mDNMT1 (731-602)
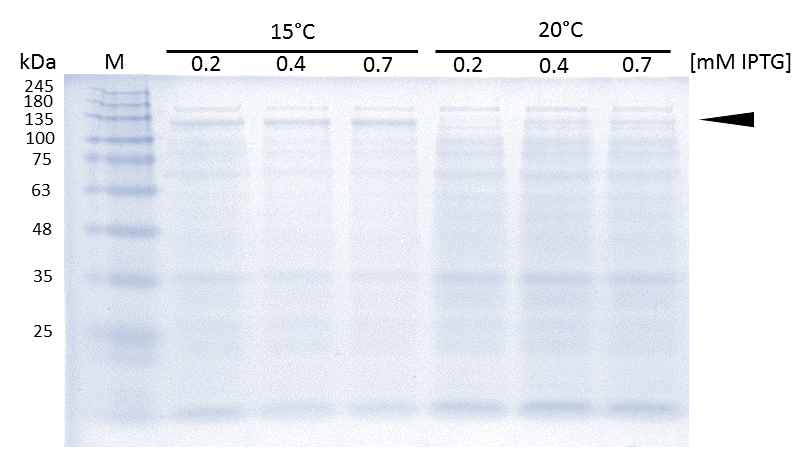
As soon as transformed Rosetta (DE3) cultures reached an OD600 of approximately 0.6, induction of linear mDNMT1 (731-1602) was perfomed over night at either 15°C or 25°C, using different concentrations of IPTG. Higest expression of DNMT1 was observed at 15°C using 0.7mM IPTG.
Before starting circularization of DNMT1, we first tested if we can actually express and purify active linear DNMT1. Since DNMT1 expression in E.coli normally results in low yields (<1mg/liter of culture) [15], we started with an optimization of the expression conditions.
In general, low temperatures and low IPTG concentrations are known to prevent the occurrence of insoluble inclusion bodies under conditions of overexpression. Thus, we tested different IPTG concentrations and temperatures during induction in order to maximize yields of active mDNMT1 (731-1602). We observed an optimal induction of our mDNMT1 construct after incubation at 15 °C using 0.7 mM IPTG (Figure 5).
After successful expression, the next critical step for recombinant protein purification is protein extraction. The lysis method has to be efficient but still gentle, especially when dealing with sensitive and low expressed proteins such as DNMT1. In order to optimize protein extraction we therefore compared cell lysis by sonification with disruption via French press (Figure 6A and 6B). Due to the low overall yield, mDNMT1 (721-1602) does not appear as a prominent band on the Coomassie-stained gel as normally expected for overexpressed recombinant proteins (Figure 6) . Since the efficiency of DNMT1 extraction could not be estimated from the Coomassie-stained gel, a western blot was performed. French press efficiently extracts soluble mDNMT1 (731-1602) whereas sonification leads to mDNMT1 accumulation in the pellet after ultracentrifugation (see Figure 6B, SN and pellet for sonification ). Indeed, the disruption of E.coli using French press has been reported to preserve the biological activity of susceptible proteins more efficiently than sonication [18]. Therefore we used the French press for further extraction of linear as well as circular mDNMT1 (731-1602).
In the western blot, several other peptides with lower molecular weight were detected. These peptides possibly result from incomplete translation or degradation processes, since the construct used for initial mDNMT1 (731-1602) expression and purification (Figure 6) comprised an N-terminal hexa-histidine tag. To minimize the amount of incomplete protein products that would be enriched in the following purification, we optimized the DNMT1 construct by shifting the His-tag to the C-terminus.
Further purification of the protein using a His-trap resulted in successful enrichment of mDNMT1f(731-1602) (Figure 6C). Corresponding eluates were pooled and further concentrated for use in functional assays.
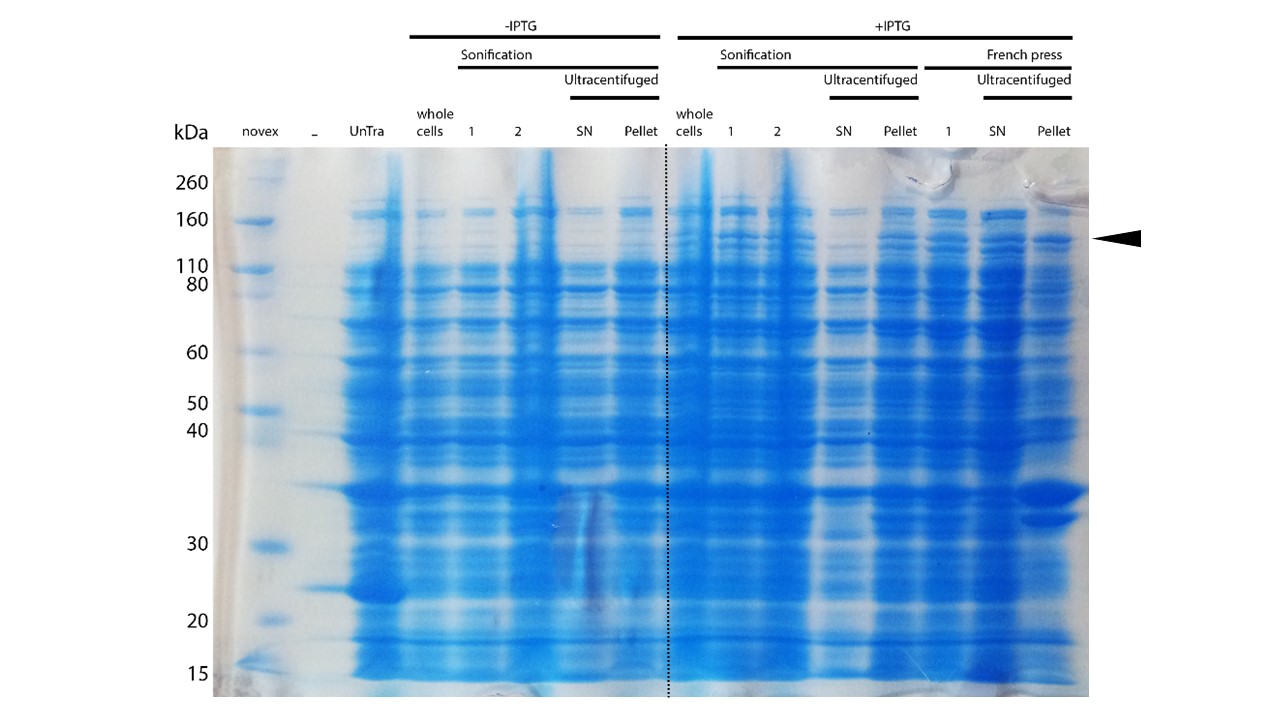
mDNMT1 (731-1602) expression in Rosetta (DE3) was performed over night at 15°C. Subsequently, the bacteria were lysed by sonification or French press and ultracentrifuged. After each step, samples were collected for analysis by SDS-PAGE. The resulting protein-membrane pellets and lysate supernatants were analyzed by SDS-PAGE. (Untransformed negative control (UnTra), intervals of cell disruption (1, 2), supernatant (SN)).
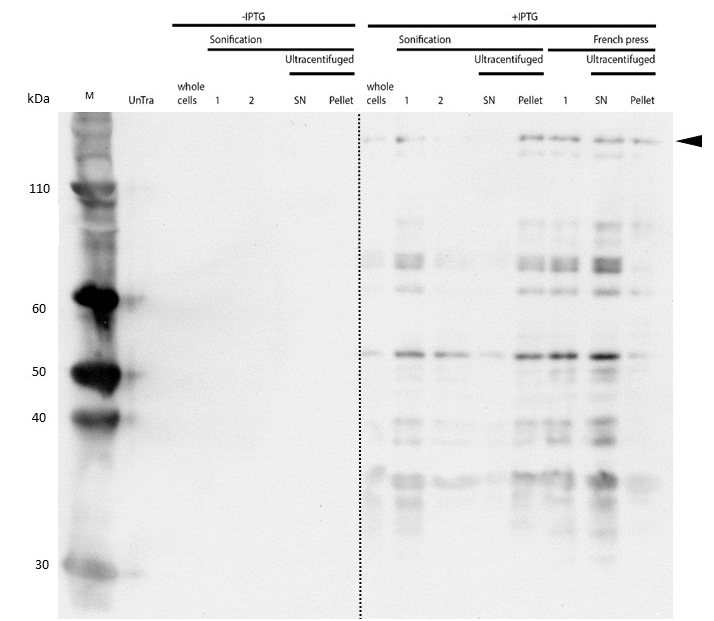
Western Blot of lysates was performed with an anti-hexa-histidine antibody for specific detection of mDNMT1. Full length mDNMT1 (731-1602) is indicated with an arrow.
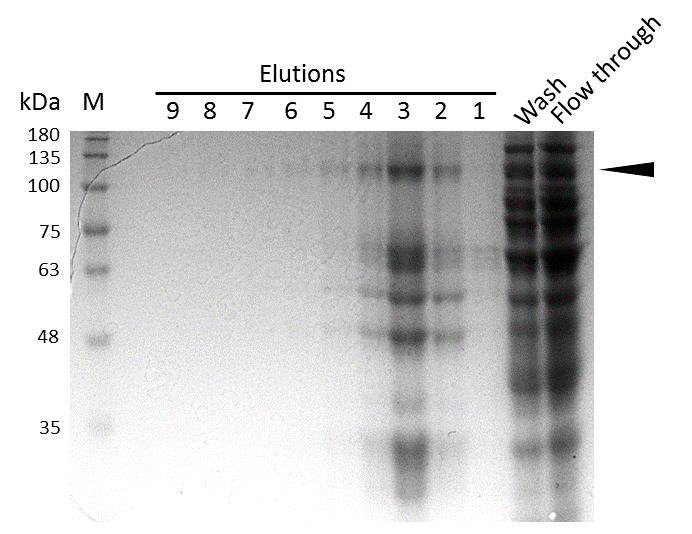
Coomassie stained SDS-PAGE showing efficient purification of linear mDNMT1 (731-1602) via His-column. Arrow indicates peptides corresponding to the molecular weight of mDNMT1 (731-1602).
Activity and specificity of linear mDNMT1 (731-1602)
After successful expression and purification of linear DNMT1, we characterized the proteins activity and specificity for hemi-methylated DNA by performing the methylation activity assay described above. To test the maintenance methylation activity of our linear DNMT1, we performed the methylation assay with different incubation times of DNMT1 (731-1602). We observe an increasing amount of completely methylated template over time (Figure 7), represented by increasing amounts of template that are unsusceptible towards SauAI cleavage.
The truncated DNMT1 (731-1602) showed maximal methylation of the template after approximately 30min. Therefore, our data agrees with efficiencies that have been reported in literature for wild type and truncated versions of mDNMT1 [15][16] .Overall, the reaction kinetics of the linear protein seem to be promising for later use in a "PCR2.0", assuming that circularization of the protein will not ablate its function.
In oder to be used in the targeted "PCR2.0" mDNMT1 needs to be highly specific for hemi-methylated sites and should not display de novo methylation activity. Therefore, we included HpaII digestion in our assay to determine de novo methylation of CpG sites. The first results prove that the purified mDNMT1 (731-1602) does not exhibit de novo methylation activity (Figure 8). Even for increasing incubation times, HpaII is not repressed which indicates absence of de novo methylation.
However, the analysis of the gel pictures is difficult since restriction efficiency of HpaII was not 100% as it can be seen from the hemimethylated control. Therefore, uncut template is possibly not resulting from unspecific methylation activity of the enzyme. Further tests, especially with the circularized protein will have to be performed to confirm these initial data.

Purified linear mDNMT1 (731-1602) was incubated with 40mer DNA template (see Figure4) for indicated times at 37°C. Subsequently the samples were digested with Sau3AI. Fragments were separated by PAGE and stained with ethidium bromide. (Homo me= homomethylated substrate, control for enzyme inhibition through methylation, Hemi me=hemimethylated substrate positive control for digest).
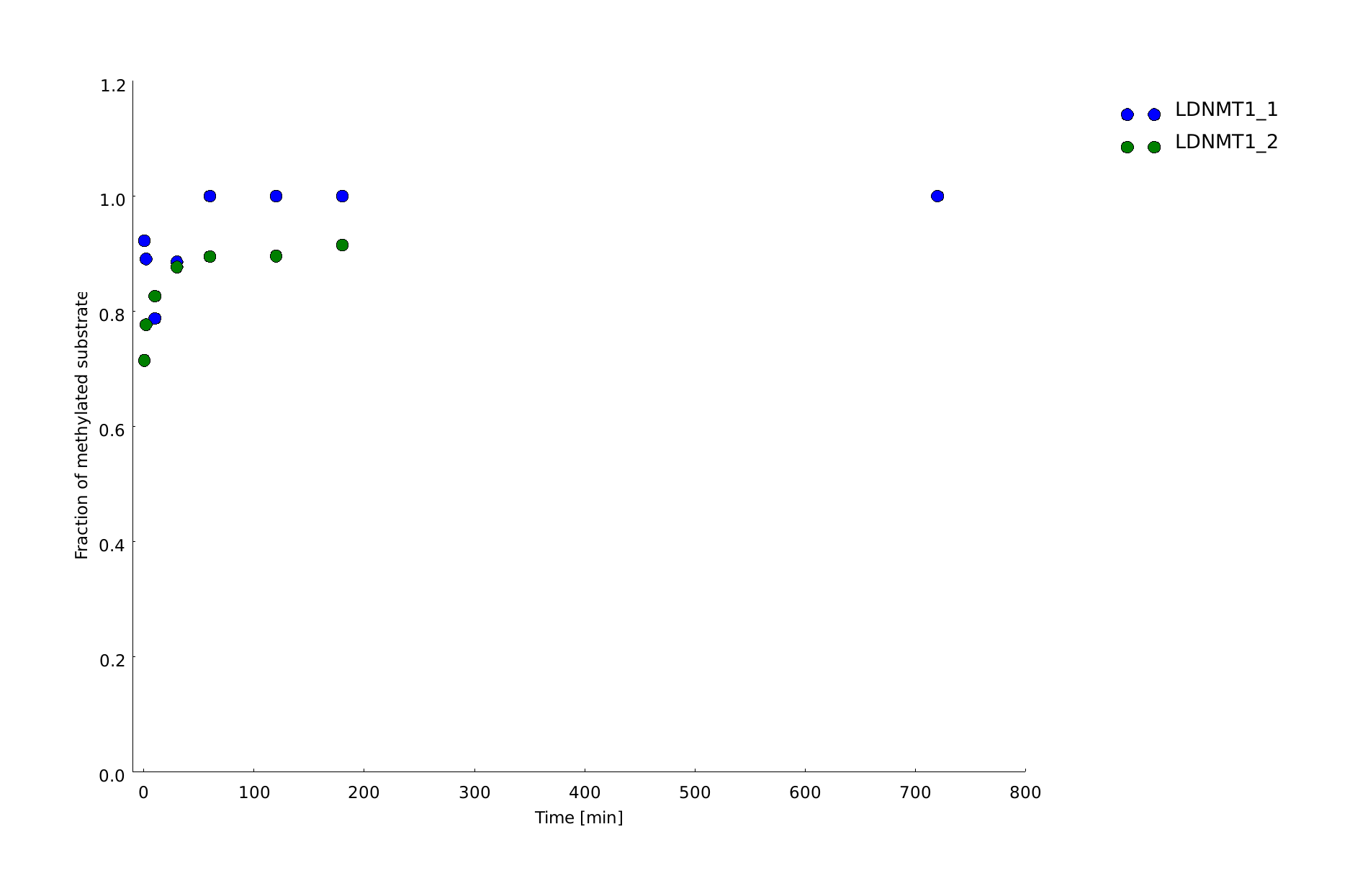
Amount of methylation was calculated by normalizing the intensity of the upper band to the overall intensity of both bands. Graph shows technical duplicate of the assay. All measurements were performed with ImageJ.
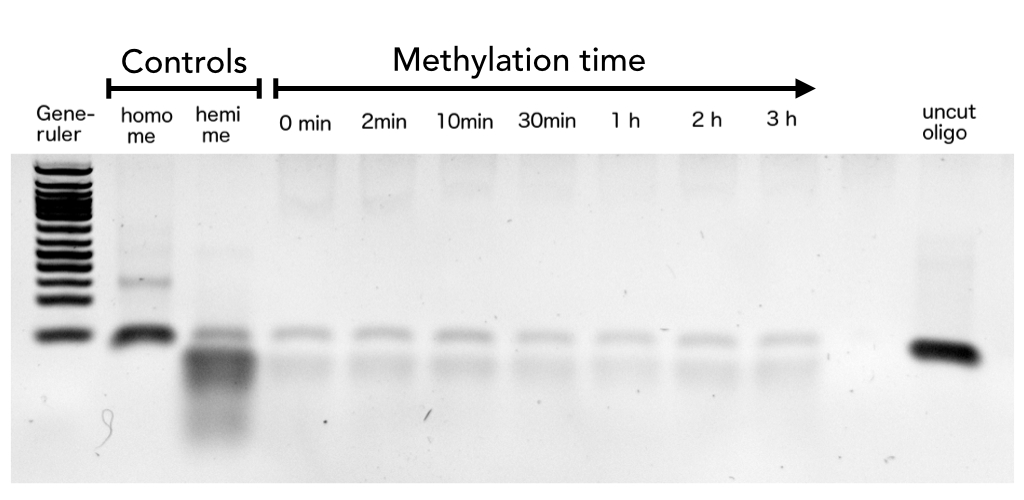
Purified linear mDNMT1 (731-1602) was incubated with 40mer DNA template (see Figure4) for indicated times at 37°C. Subsequently the samples were digested with HpaII. Fragments were separated using gel-electrophoresis and visualized with ethidiumbromid staining. (Homo me= homomethylated substrate,control for enzyme inhibition through methylation, Hemi me=hemimethylated substrate positive control for digest).
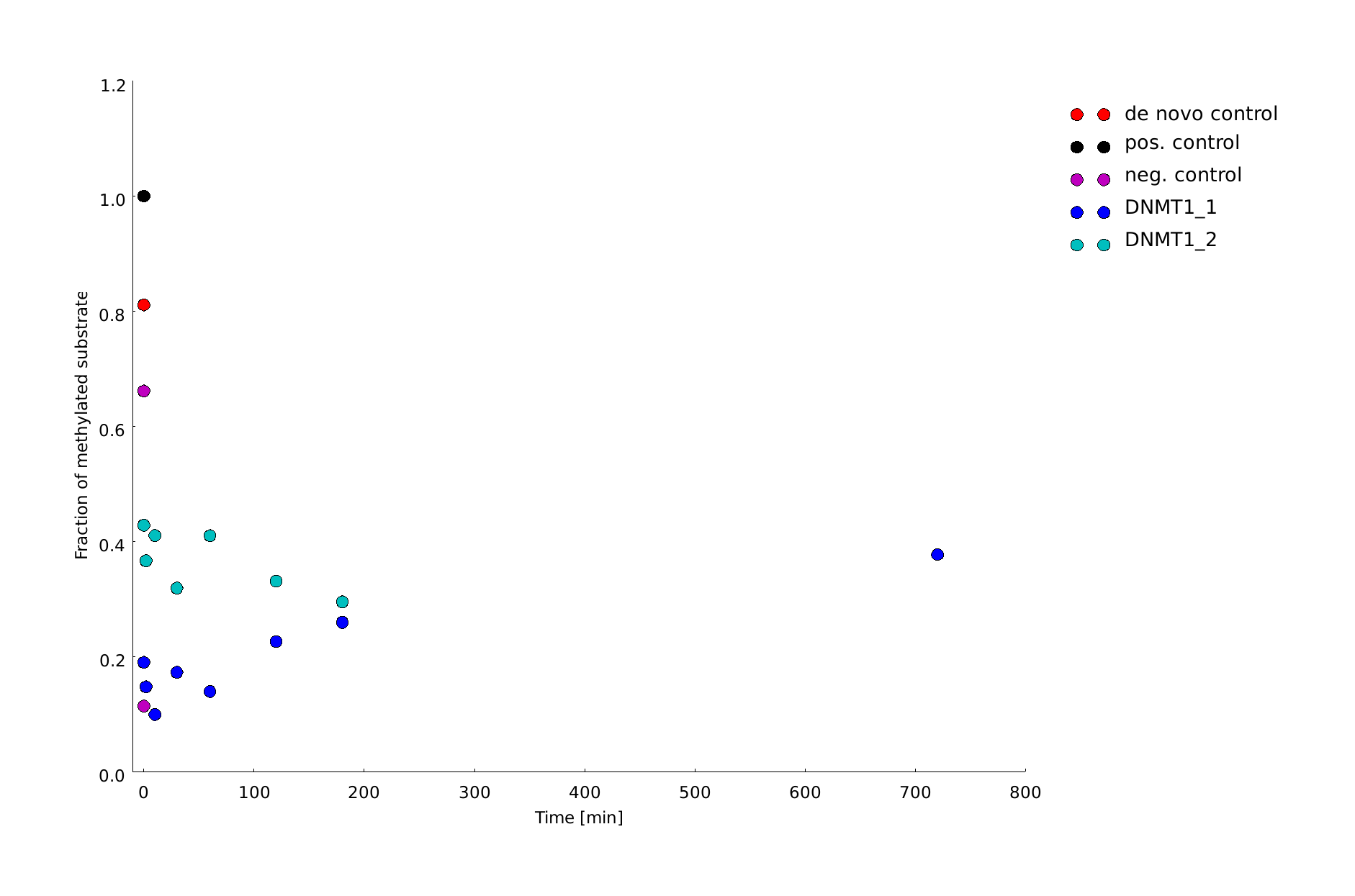
Amount of methylation was calculated by normalizing the intensity of the upper band to the overall intensity of both bands. All measurements were performed with ImageJ.
Circular mDNMT1 (731-1602) - The pivotal element of PCR2.0
Expression and purification of mDNMT1 (731-1602) for circularization
After we succeeded in purifying active and specific linear mDNMT1, we went on with cloning, expression and purification of the circular DNMTs.
Expression and purification of mDNMT1 (731-1602) for the intein as well as the sortase approach were conducted as they had been established for the linear DNMT1. Elutions from the His-Trap showed an enrichment and concentration of peptides that correlate with the expected molecular weight of the truncated mDNMT1 (731-1602) (Figure 9A and 9B). Nevertheless, purification with a His-Trap alone does not seem to exclude a variety of impurities that either result from degraded mDNMT1 that still contains a His-Tag or unspecific binding to the affinity column. Advanced protocols as they were used by Song and collegues [19] for analysis of the proteins crystal structure are therefore including several more steps. Nevertheless, additional steps of purification increase the risk of reducing the overall protein yield and activity, necessitating greater amounts of starting material. Since the active protein should already be included and efficiently enriched in our sample despite of present impurities, we continued with the functional analysis of the sample.
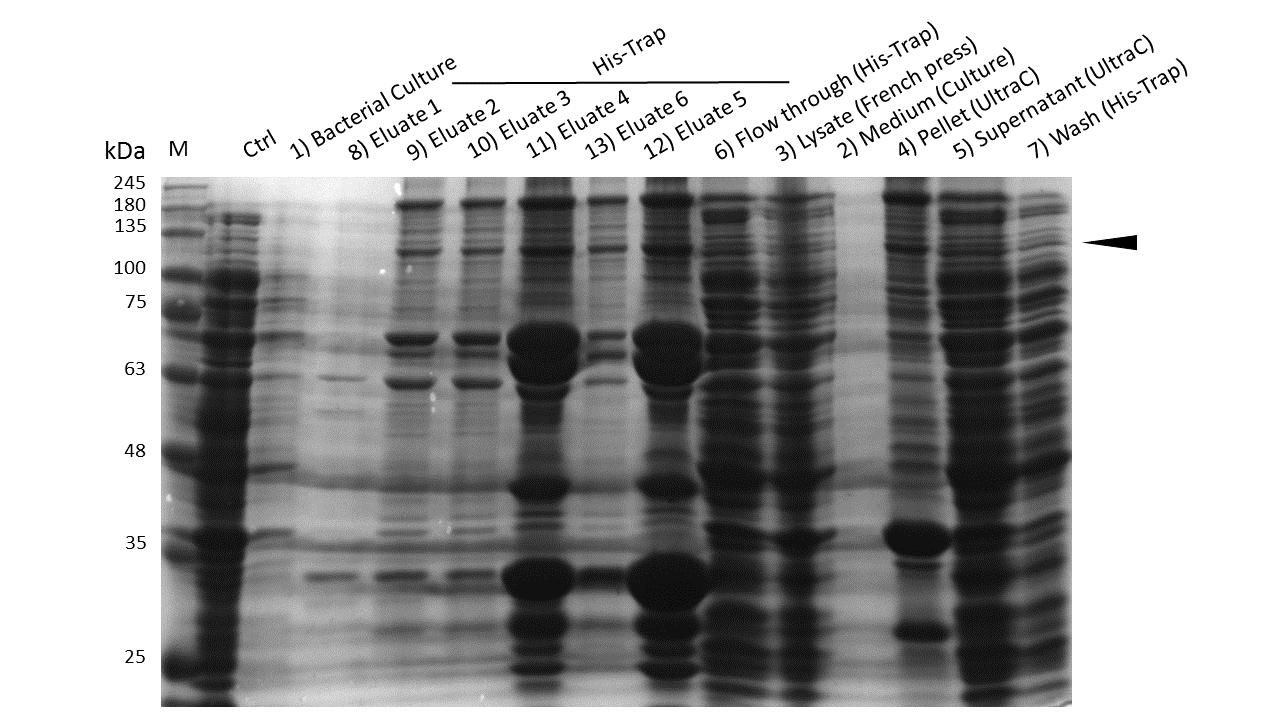
Induction of Rosetta (DE3) and purification were performed as described above. After each step, samples were collected for analysis by Coomassie-stained SDS-PAGE. Samples are numbered according to their chronology of collection. Arrow indicates mDNMT1 (731-1602).

Induction of Rosetta (DE3) and purification were performed as described above. After each step, samples were collected for analysis by Coomassie stained SDS-PAGE. Samples are numbered according to their chronology of collection. Arrow indicates mDNMT1 (731-1602).
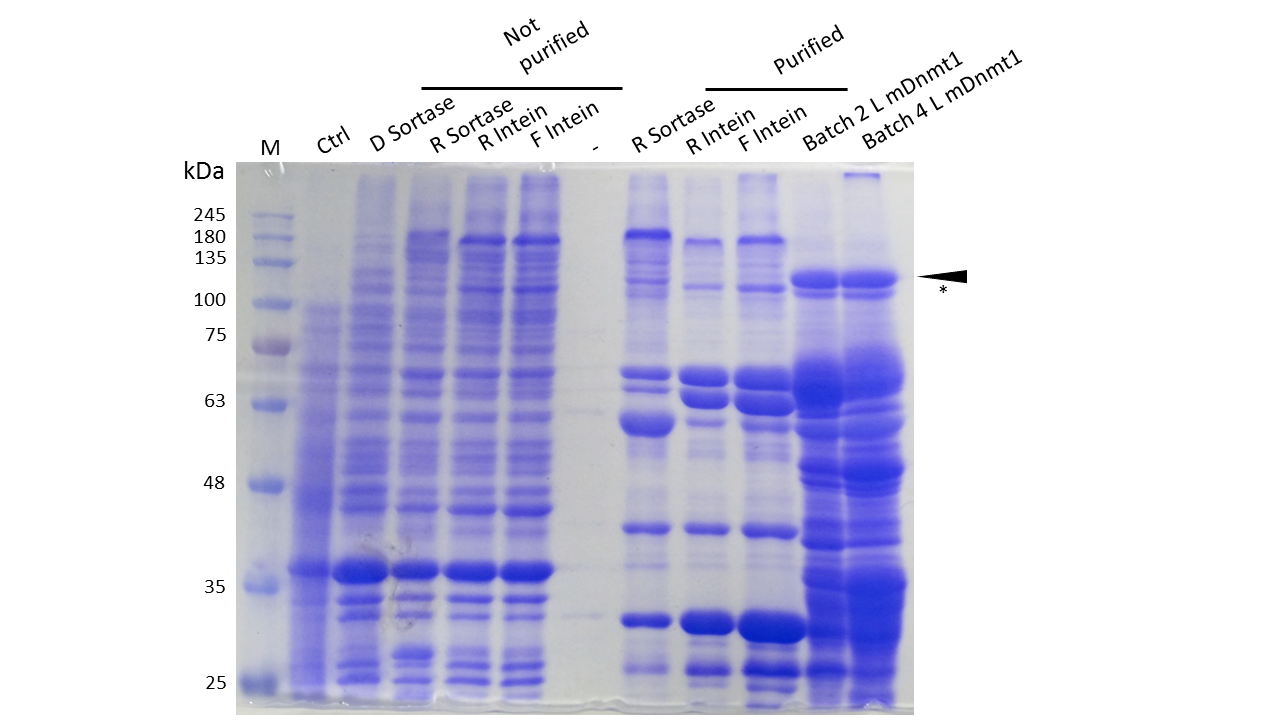
Expression and Purification of different constructs in Rosetta (DE3) was performed as described above. Unpurified as well as purified samples were collected for analysis by Coomassie-stained SDS-PAGE.(Ctrl indicates the untransformed control. D= no linker, R= Rigid linker, F=Flexible linker, L= linear DNMT1. Arrow indicates peptides with molecular weight corresponding to mDNMT1 (731-1602). Asterix indicates possibly spliced Intein constructs).
Our approach of heat-stabilization includes the testing of different linkers that had been calculated by our CRAUT Linker Software. We tested a flexible linker, consisting mostly of glycines and serines and a rigid linker, which was introduced to maximize stabilization of the protein through cicularization. Purification of the mDNMT1 (731-1602) variants with different linkers for circularization was successful (Figure 10), but the yields were lower compared to different batches of linear mDNMT1 (731-1602) that had been produced in a similar way.
Interestingly, a shift between mDNMT1 (731-1602) expressed from the intein construct compared to the sortase or linear versions was detected. Since protein circularization via inteins is an autocatalytic process that takes place inside the bacteria, this shift could possibly result from successful splicing activity of the inteins. The resulting product lacks Smt3 and is therefore approximately 30kDa smaller than its precursor (Figure 10, indicated by an asterix). Since intein splicing likely occurred, flexible- and rigid-linked-DNMT1 could be circularized.
To prove circularization by inteins, we cooperated with the core facility "Protein Analysis" of the German Cancer Research Center (DKFZ). In the facility, circularized and linear DNMT1 were extracted from a provided gel, digested with trypsin and analyzed by ESI mass spectrometry. Unfortunately, we do not have the final results yet but are confident to be able to present them at the jamboree.
In contrast to the intein DNMT1 construct, mDNMT1 (731-1602) with sortase recognition sequences needs to be further processed before a circular protein is obtained. Unfortunately, TEV cleavage of the purified protein, which is necessary for subsequent circularization with sortase was not very efficient. Therefore sortase treatment did not yield in a detectable amount of processed mDNMT1 (731-1602). Therefore, we fully concentrated on the promising intein approach for further mDNMT1 (731-1602) circularization.
Thermal stability of circular mDNMT1s(731-1602)
In our final assay we compared the effect of a PCR-relevant heat shock on the activity of linear and circular DNMT1s. Thereby, we wanted to examine the ability of circular DNMT1 to withstand higher temperatures and the feasibility of a PCR 2.0 using circularized DNMT1. Linear DNMT1 (L) as well as rigid linker (RC) and flexible linker-containing (FC) DNMT1s were exposed for 5s to temperatures between 55°C and 72°C that would also be reached in a PCR and subsequently analyzed for methylation activity.
In the gel picture (Figure 11A and B) a darker upper band in the FC sample compared to the linear DNMT1 sample indicates higher methylation activity of circular DNMT1 with flexible Linker at all temperatures Quantification of the band intensities using Image J shows that activity of circular DNMT1 with the flexible linker (black) is exceeding the linear and circular with rigid linker at least by a factor of 2. Surprisingly, the methylation activity of circular DNMT1 with a flexible linker seems to increase with higher temperatures.
For significant evaluation, more data has to be collected. When applying Michael Menten kinetics, the methylation reaction for the controls already approaches saturation. Therefore, the normalization to the original activity might be misleading. For further improvement of the assay, less volume of DNMT1 has to be taken for the enzyme reaction.

Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 55°C and 60°C.
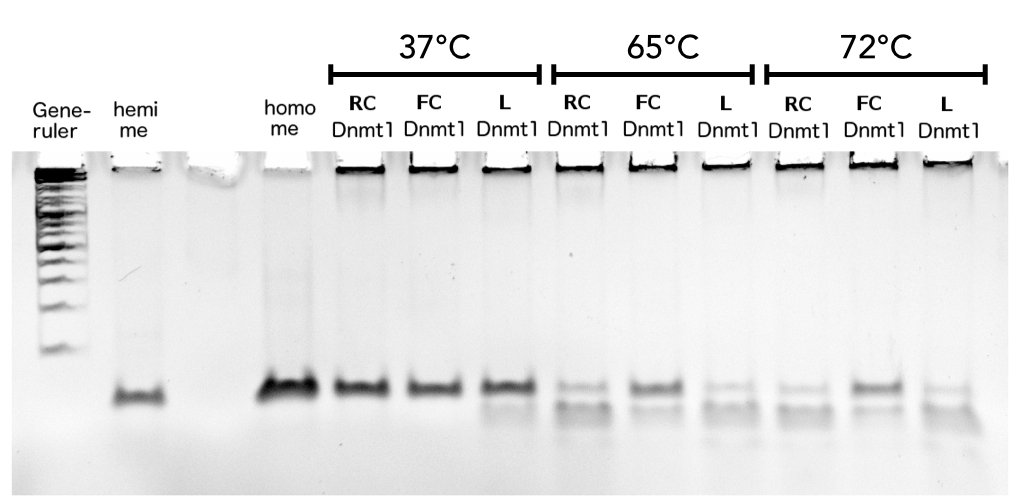
Circular DNMT1 with rigid linker (RC), circular DNMT1 with flexible linker (FC) and linear DNMT1 were heat shocked for 5 sec at 65°C and 72°C.Methylation after one hour was measured. The dark upper band for FC indicates higher methylation activity.
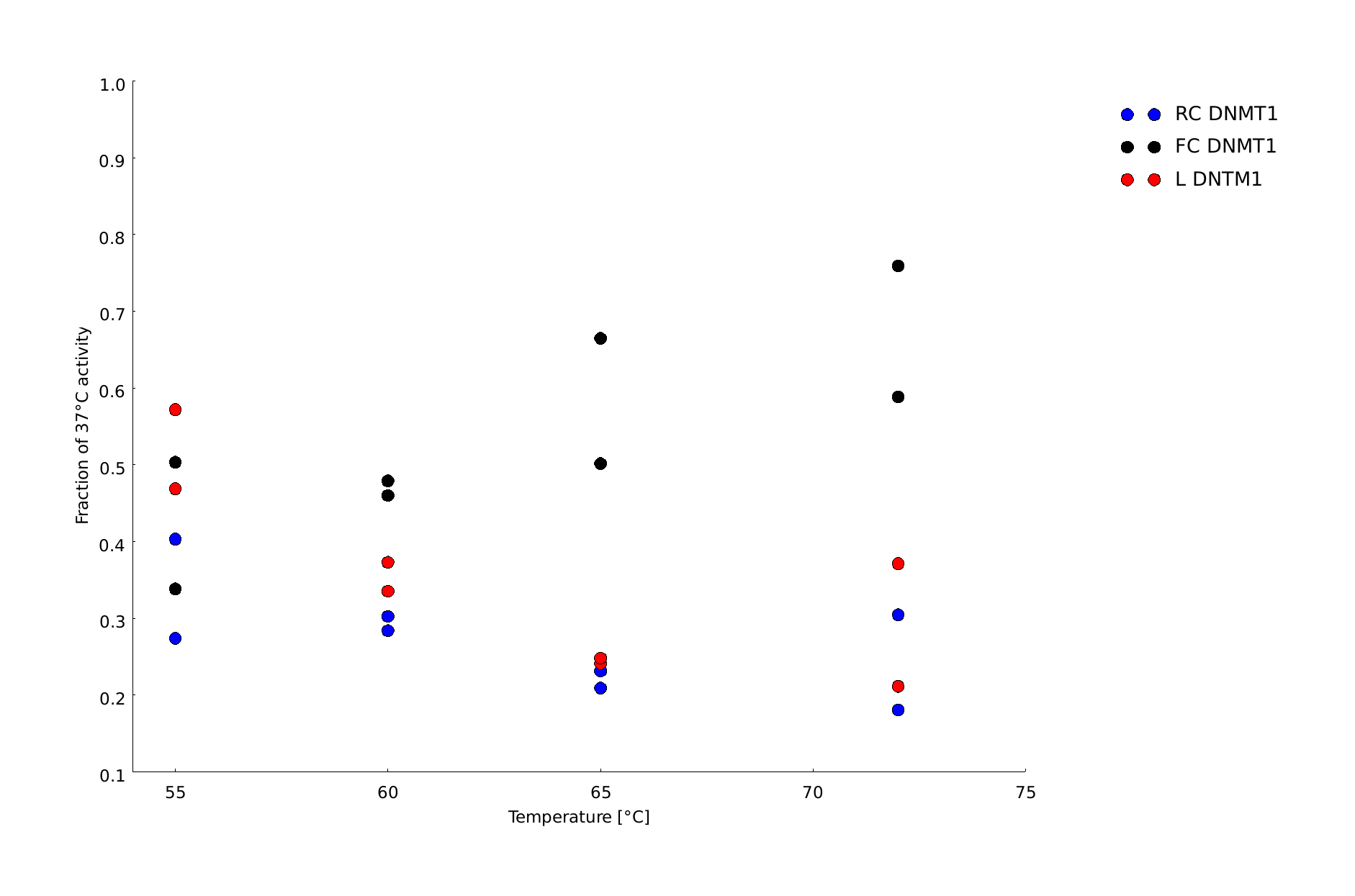
Circular DNMT1 with rigid linker (RC) in blue, circular DNMT1 with flexible linker (FC) in red and linear DNMT1 (L) in blue over a temperature range from 55°C to 72°C.
Outlook
Our next steps until the jamboree will be to prove efficient linkage of N- and C- terminus for our circularized mDNMT1 (731-1602) via mass spectrometry. Further methylation activity assays with circularized protein are planned to validate significance of the obtained data. Moreover, we are looking forward to use our heat-stable mDNMT1 (731-1602) in the first ”PCR2.0”.
Discussion
Achievements and impact
Taken together, we could show that intein-mediated circularization of mDNMT1 (731-1602) conceivably increased heat stability of the enzyme without impeding specific methylation activity. We proved that circularization with inteins is an easy method to increase heat stability of complex and sensitive proteins like mDNMT1 (731-1602). In comparison, other methods used for protein stabilization such as introduction of disulfide bonds or hydrophobic amino acids, require sophisticated structure analysis or non-physiological reaction conditions [20][21]. In fact, mDNMT1 (731-1602) is the largest protein that has ever been circularized so far and even more might follow.
Our CRAUT Linker Software was essential to achieve circularization of mDNMT1 (731-1602) while preserving its natural function. In first experiments, the two designed linker compositions resulted in divergent heat stabilities of the protein. Our software successfully narrowed down the infinite amount of possibilities to circularize mDNMT1 (731-1602). Further experimental data of enzyme-specific linker variants will help to further improve the software predictions. Circularized mDNMT1 (731-1602) represents a valuable tool for the realization of a methylation maintaining ”PCR2.0”. This foundational advance aims to enable broad functional analysis of DNA methylation patterns without prior knowledge on methylation frequency or dependency on other methods. For the detection of DNA methylation using bisulfite sequencing, handling of limited amounts of primary material still represents a major challenge:
| “Future developments will undoubtedly allow information to be recovered from most genomic CpGs, the key being the ability to amplify DNA before bisulfite conversion. The capacity to capture the DNA methylome from individual cells will be critical for a full understandingof early embryonic development, cancer progression and generation of induced pluripotent stem cells.” - Smallwood et al., Nat. Methods (20014) [29] |
Approaches including single molecule sequencing have been reported, but have not been established in an affordable and efficient manner yet [27]. Furthermore, these methods are limited to the detection of methylation patterns and do not allow functional analysis without complete analysis and cost-intensive synthesis. Therefore, our ”PCR2.0” aims to revolutionize the field of epigenetics by linking complex methylation patterns to their actual function. In vitro as well as in vivo experiments, requiring large amounts of methylated DNA will be feasible by using our ”PCR2.0”. Those include for example the detection of DNA methylation binding proteins using Chromatin Immunoprecipitation or Electrophoretic Mobility Shift Assays. Further functional studies could investigate cellular regulation upon transfection with methylated template.
A very interesting application for the pharmaceutical use of our "PCR2.0" could be the large scale production of methylated gene therapy vectors. Since methylation of gene transfer vectors has been reported to extend transgene product expression by lowering cellular immunogenicity, the efficiency gene therapies might be improved with our method.[22]
Considerations for further PCR2.0 development
Since significance of the obtained methylation activity data still has to be proven in biological replicates, we could not yet precisely quantify the effect of DNMT1 stabilization by circularization. Therefore further steps in addition to the circularization of mDNMT1 (731-1602) could be necessary to obtain the required heat stability and specificity for our methylation maintaining "PCR 2.0". Those for example include further protein stabilization by enhancing the hydrophobicity of the protein core or by introducing direct links to limit possible conformations [23].
Nevertheless, it is unlikely that mDNMT1 (731-1602) can be stabilized in a way to bear temperatures of 95°C, which are essential for conventional PCR. Therefore, another upcoming topic is the method of low temperature PCRs that exploit the function of helicases to segregate DNA strands. Even though these PCRs can be performed at lower temperatures than conventional PCRs, none of them has reached widespread establishment yet. Main reasons are that isothermal PCRs require either too long incubation times per cycle [24] or still exceed physiological temperatures of most enzymes, introducing new candidates for circularization... [25]
Despite enduring activity of mDNMT1 (731-1602) at high temperatures, the heat-stability of its cofactors also plays a crucial role for establishing a ”PCR 2.0”. Here, a still challenging factor is methyl-donor S-adenosyl methionine (SAM) that exhibits only low stability at high temperatures [30]. Nevertheless, addition of the quite cheap methyl-donor after each cycle of replication could circumvent this problem until a more stable variant of the cofactor is available.
If there are occurring problems with the specificity of the enzyme at higher temperatures which have not been assessable from our data, the “PCR 2.0” could be complemented with enzymes such as UHRF1, that have been reported to increase the specificity of mDNMT1 (731-1602) [31]. Wild type DNMT1 is known to be associated to the replication complex for direction of its activity. Design of an appropriate Polymerase-DNMT-fusion protein could imitate this connection and therefore further reduce stochastic methylation events [26].
Overall, the iGEM Team Heidelberg has achieved important milestones in establishing a methylation maintaining "PCR 2.0" by expressing and purifying the largest circular protein ever. Despite of its use in a "PCR 2.0", the stabilized mDNMT1 (731-1602) can also be used in other methylation assays.
References
[1] Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–70 (2008).
[2] Watanabe, M., Yuzawa, H., Handa, N., & Kobayashi, I.. Hyperthermophilic DNA methyltransferase M.PabI from the archaeon Pyrococcus abyssi. Applied and Environmental Microbiology, 72(8), 5367–75 (2006).
[3] Vieille, C. & Zeikus, G. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65, 1–43 (2001).
[4] Antos, J. M., Popp, M. W.-L., Ernst, R., Chew, G.-L., Spooner, E., & Ploegh, H. L.. A straight path to circular proteins. The Journal of Biological Chemistry, 284(23), 16028–36 (2009).
[5] Tam, J. P. & Wong, C. T. T. Chemical synthesis of circular proteins. J. Biol. Chem. 287, 27020–5 (2012).
[6] Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005).
[7] Jaenisch, R. & Bird, A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33 Suppl, 245–54 (2003).
[8] Ehrlich, M.. Amount and distribution of 5-methycytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 10: 2709–2721 (1982).
[9] Lister, R. & Ecker, J. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 959–966 (2009).
[10] Bird, A.. DNA methylation patterns and epigenetic memory. Genes & Development 16, 6-21 (2002).
[11] Jaenisch, R., and Bird, A.. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33, 245-254 (2003).
[12] Weichenhan, D., and Plass, C.. The evolving epigenome. Human Molecular Genetics 22, R1 (2013).
[13] Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005).
[14] Jurkowska, R. Z., Jurkowski, T. P. & Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 12, 206–22 (2011).
[15] Song, J., Teplova, M., Ishibe-Murakami, S. & Patel, D. J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 335, 709–12 (2012).
[16] Bashtrykov, P. et al. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem. Biol. 19, 572–8 (2012).
[17] Lee, C.-D., Sun, H.-C., Hu, S.-M., Chiu, C.-F., Homhuan, A., Liang, S.-M., Wang, T.-F. An improved SUMO fusion protein system for effective production of native proteins. Protein Science : A Publication of the Protein Society, 17(7), 1241–8 (2008).
[18] Benov, L. & Al-ibraheem, J. Disrupting Escherichia coli : A Comparison of Methods. 35, 428–431 (2002).
[19] Song, J., Rechkoblit, O., Bestor, T. H. & Patel, D. J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–40 (2011).
[20] Betz, S. F. Disulfide bonds and the stability of globular proteins. 1551–1558 (1993).
[21] Bulaj, G. Formation of disulfide bonds in proteins and peptides. Biotechnol. Adv. 23, 87–92 (2005).
[22] Reyes-Sandoval, a & Ertl, H. C. J. CpG methylation of a plasmid vector results in extended transgene product expression by circumventing induction of immune responses. Mol. Ther. 9, 249–61 (2004).
[23] Vieille, C. & Zeikus, G. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65, 1–43 (2001).
[24] Vincent, M., Xu, Y. & Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 5, 795–800 (2004).
[25] Xu, G. et al. Cross priming amplification: mechanism and optimization for isothermal DNA amplification. Sci. Rep. 2, 246 (2012).
[26] Vilkaitis, G., Suetake, I., Klimasauskas, S. & Tajima, S. Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J. Biol. Chem. 280, 64–72 (2005).
[27] Krueger, F., Kreck, B., Franke, A. & Andrews, S. R. DNA methylome analysis using short bisulfite sequencing data. Nat. Methods 9, 145–51 (2012).
[28] Smith, Z. D. & Meissner, A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–20 (2013).
[29] Smallwood, S. a et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 6–11 (2014).
[30] Matos, J. & Wong, C. S-adenosylmethionine: Stability and stabilization. Bioorg. Chem. 80, 71–80 (1987).
[31] Bashtrykov, P., Jankevicius, G., Jurkowska, R. Z., Ragozin, S. & Jeltsch, A. The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J. Biol. Chem. 289, 4106–15 (2014).