 "
"
Team:Heidelberg/pages/Linker Screening
From 2014.igem.org
We developed a modeling approach to design linkers that can circularize proteins and that may confer some rigidity in particular after heat shock. The general concept of the approach can be found in the Software and the Modeling sections
The experimental work presented in this section has two main goals. The first one was testing the validity of the idea: Can rigid linkers with angles indeed provide heat stability to a protein better than flexible linkers? And the second one was to measure this stabilization depending on the properties of the linkers to calibrate the Linker Software. As already seen in the software part, these properties are the total length of the linker, the angles between consecutive alpha helices, the regions that the linker passes and the distance from the proteins surface. As we had no a priori knowledge on the contribution of those different properties for the heat stability of the protein, we performed an extensive linker screening on the lambda phage lysozyme. The linkers checked were predicted to different groups by the software: Very good, somehow good, maybe working, bad and too short.
Contents |
Introduction
Circularization is a narrow path between gaining heat-stability and loosing function due to deformation.
The choice of the protein to perform the linker screen was constrained by different requirements. First, it needed to be easily and fast expressed in E.coli. We needed to be able to measure its functionality without purification and in an easy, fast, cheap and reliable way. It needed to be known for having a certain stability at high temperature with, at the same time, a loss of functionality so that the heat stabilization could be tested. Finally, the major constrain for the choise was the structure of the protein. First, we needed a cristal structure of the complete protein at high resolution. Second, the ends should be separated by a distance of 15 to 30 Angströms, so that the rigid linkers containing alpha helices would be relevant.
Lysozymes are well characterized enzymes that are able to digest the peptidoglycans that form the bacterial wall. This process is performed by many different species for different applications, including antibacterial defense by plants and animals [1] or bacterial penetration by viruses [2]. On top, lysozyme is applied in different fields of biotechnology and medicine. It is notably one of the most important proteins in food preservation and is produced in the 100 tons scale a year. We anticipated that the lysozyme of the bacteriophage lambda could reasonably fulfill the requirements for our linker screen. Its crystal structure is known [3] and the ends are 27.3 Angströms apart (figure 1).
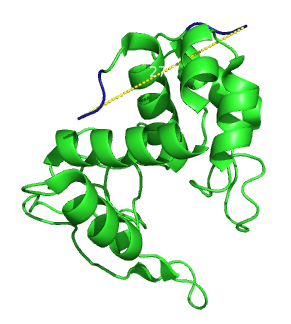
Secondary structure of lambda lysozyme. The distance between the two ends is 27.3 Angströms.
Main Results
We could clone and express the lysozyme of bacteriophage lambda and its fusion to different linkers (table 1) chosen to calibrate our linker software. We used lyophilized Micrococcus lysodeikticus as substrate to measure lysozyme activity and we established a heat shock assay to measure the heat-stability of the enzyme. We observed that while lysozyme shows a clear activity at 37°C, it is gradually lost when the enzyme is pretreated at higher temperatures up to 57°C. Thanks to our measurement of the kinetics of substrate processing, we could extract information on the amount of active enzyme at different heat shock temperature and with different linkers by modeling this activity. The following figure shows the substrate processing measured from the decrease of optical density of the M. lysodeikticus for the different temperatures of heat shock and the different linkers.
Experimental Process
Cloning of lysozyme fused to linkers
To get the circular form of lambda lysozyme we used the autocatalytic function of the intein NpuDnaE, an often used and well-described split-intein. Lambda lysozyme was cloned in our new standard of circularization, described in the article about Circularization.
Expression of linker lysozymes and test of circulatization
With a Coomassie Gel and a Western Blot we tested the expressed construct on circularization. Circular proteins run faster in a gel than proteins of the same length because of their structure. So you can see a shift on the Western Blot between the linear and the circular form of a protein (figure 2).
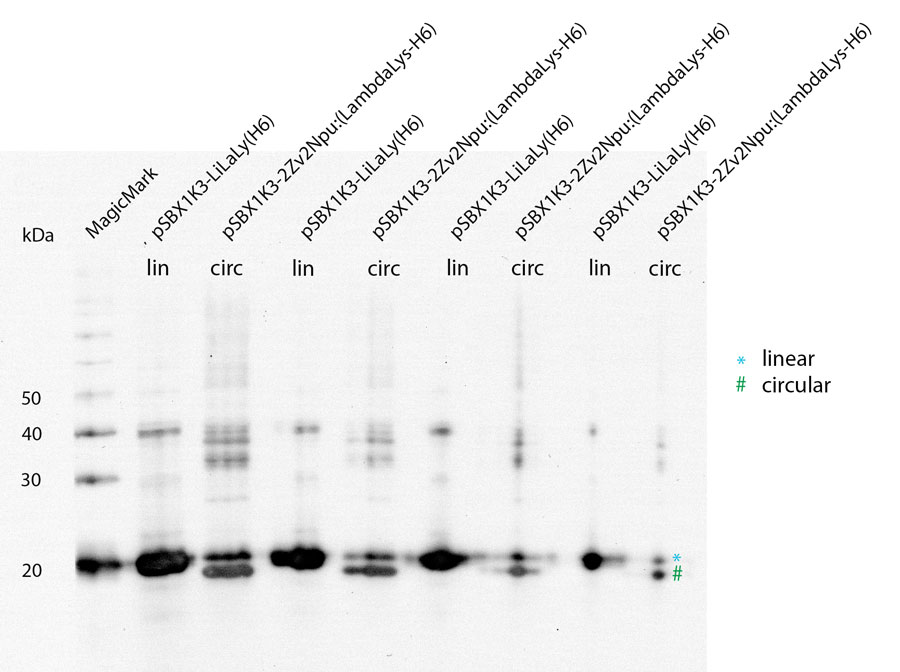
To test if lambda lysozyme is circular after the splicing reaction of the intein Npu Dna E we did a Wetern Blot with Penta·His Antibody. There is a clear shift between linear and circular samples.
Because of the high expression level, tested with a Coomassie Gel (figure 3), we did not purify the lysozyme constructs and used non-purified protein mix. On the gel in figure 3 you can see that there is a difference between the expression level of the different constructs. But all constructs are well enough expressed to work with them.
The linker screening was performed with ten different linkers. We wanted to show that different linkers have different effects on the thermostability and function of a protein, as claimed by our ###Link. The linker software calculated linkers classified according to fitness as great, bad, short and proper linker. The length of a linker has an significant effect on folding structure of proteins.
Development of assay to measure lysozyme activity
To test and quantify the differences in thermo stability of the linear and several circular forms with different linkers of lambda lysozyme we needed an assay that is simple and reproducible enough. To have a comparison and a great check on functionality we tested two different methods to measure lysozyme activity and establish the range of temperature to test the heat stability of the enzyme. Lysozyme of the bacteriophage lambda like the other lysozymes cleaves glycosidic bonds between 1,4-beta-linkages between N-acetylmuramic acid (NAM) and N-acetyl-D-glucosamine residues (NAG) in peptidoglycan and between N-acetyl-D-glucosamine residues in. As a really small protein built of only 158 amino acids its mechanism is different from other lysozymes: the hydroxyl function OH on the C6 of the muramic acid is the nucleophile on cleavage of the muropeptide instead of the more common water molecule. So the protein is a transglycosidase, not a hydrolase [3]. The scientific background of the first assay was the capacity of lambda lysozyme to degradate the cell wall of Micrococcus lysodeikticus, a gram-positive bacterium often used as a lysozyme substrate [4]. The rate of lysis of Micrococcus lysodeikticus was measured by a change of OD of the substrate solution at 600nm. Using 96-well plates we have the possibility to test on several constructs within one assay.
The basis of the second assay was the labeling of peptidoglycan with fluorescein isothiocyanate (FITC), an amine-reactive derivative of fluorescein dye. When the FITC-labeled substrate was subjected to the enzyme digestion, an increase of fluorescence intensity and a decrease of fluorescence polarization value should be observable [5].
Materials and Methods
Results
Discussion
References
[1] Callewaert, L. & Michiels, C. W. Lysozymes in the animal kingdom. J. Biosci. 35, 127–160 (2010).
[2] Evrard, C., Fastrez, J. & Declercq, J. P. Crystal structure of the lysozyme from bacteriophage lambda and its relationship with V and C-type lysozymes. J. Mol. Biol. 276, 151–64 (1998).
[3] Callewaert, L. et al. Purification of Ivy, a lysozyme inhibitor from Escherichia coli, and characterisation of its specificity for various lysozymes. Enzyme Microb. Technol. 37, 205–211 (2005).
[4]
[5] Maeda, H. A New Lysozyme Assay Based on Fluorescence Polarization or Fluorescence Intensity Utilizing a Fluorescent Peptidoglycan Substrate 1. 1191, 1185–1191 (1980).