 "
"
Team:ETH Zurich/blog
From 2014.igem.org

{{{1}}}
Blog
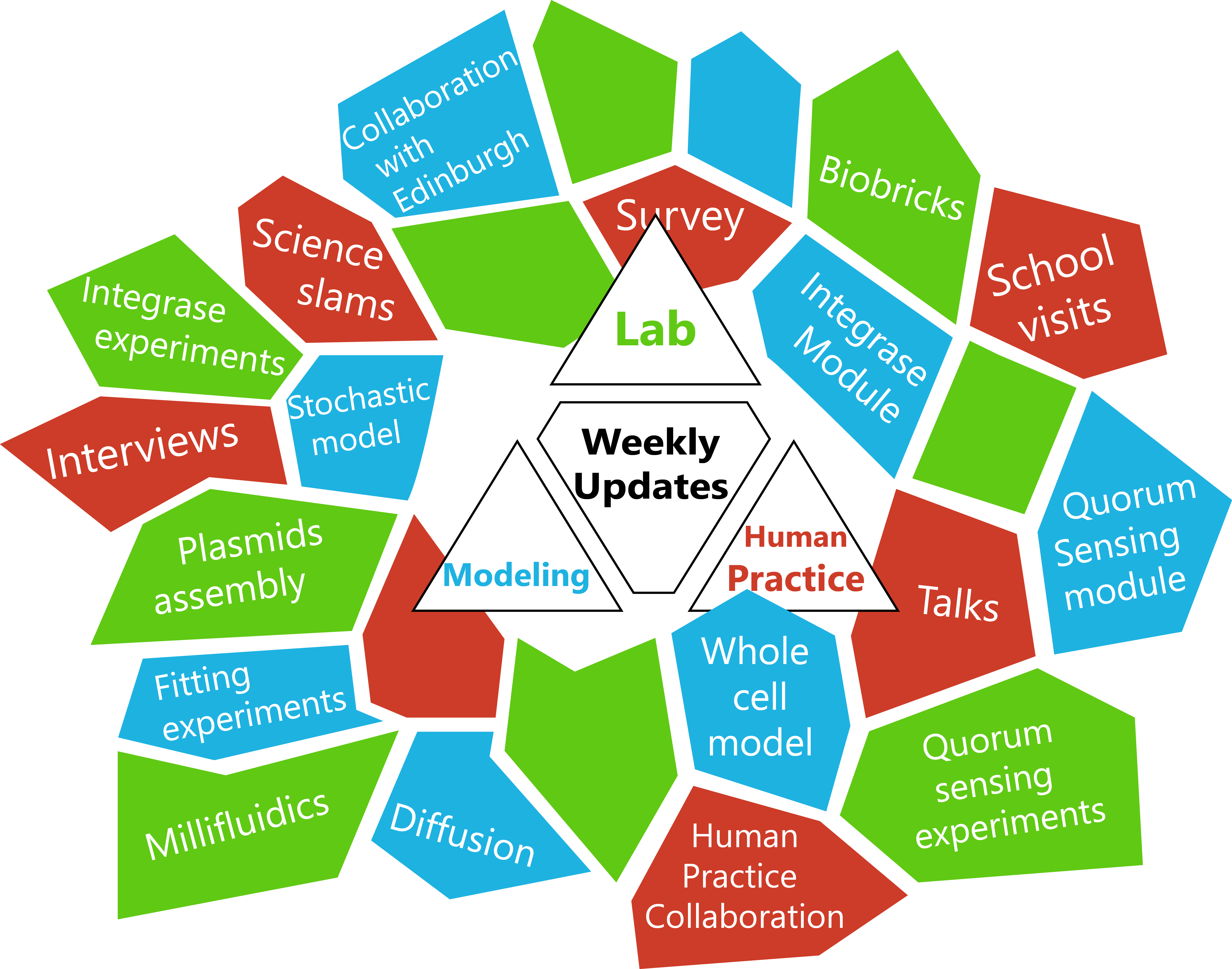
title of article
Preparation of DNA from iGEM kit
Sequencing, Transformation, antibiotic stock preparation
Miniprep
Team:ETH Zurich/labblog/20140711
Team:ETH Zurich/labblog/20140710
Week 7: Human practice planning, Plasmid assembly running, Logo design
Wednesday, July 9th
- We have a logo !

- In the lab, we did some :
- Plasmid preparation
- Sequencing
- Digests
- Purification of backbone fragments needed for GA
- On the modeling side, we tried to estimate integrase parameters from the paper from Bonnet et al. [9], more particularly with the figure S4 :

We are trying several strategies : minimization of the error function and Markov Chain Monte Carlo.
- We have a precise plan for our human practice project. We would like to study the emergence of complexity in many different fields and investigate how people deal with it. We will do this with interviews of experts in different fields, and with a survey for a wider outreach. We will also reach younger people by organizing talks in schools. We will finally give our own advice on the question by writing an essay on the subject and linking it to our experience with Mosaicoli.
Digest and miniprep
Tuesday, July 8th
Optimizing Restriction Endonuclease Reactions
Digest of 291 and 561
291: (2x)
1μL AscI
1μL FseI
0.5μL PvuI
0.5μL AseI
20 μL DNA 291
5μL Cut Smart Buffer
fill up with 22 μL H2O to 50μL
dephosphorylation of 181 and 271, cleanup with PCR clean-up system, gel electrophoresis
Gel electrophoresis
0.5 g Agarose in 50 mL TAE (0.5x), heat in microwave to dissolve
let solution cool down to approximately 50 °C, add 5 μL peqGREEN, mix
pour solution in tray, use appropriate comb
add 10μL loading dye (6x) to samples
fill samples and ladder (10 μL) in wells
run gel at 135 V for 50min


Miniprep of 35 and 36
Digest and miniprep
Monday, July 7th
Optimizing Restriction Endonuclease Reactions
Digest of 291 and 561
291: (2x)
1μL AscI
1μL FseI
0.5μL PvuI
0.5μL AseI
20 μL DNA 291
5μL Cut Smart Buffer
fill up with 22 μL H2O to 50μL
Miniprep of 35 and 36
Miniprep
Sunday, July 6th
Miniprep with bacteria cultures containing 271, 291, 181 and 30
Addition of 200 μL Resuspension solution to culture pellet, vortex, addition of 200 μL Lysis solution
After ca. 5 min addition of 350 μL Neutralization solution, centrifuge samples for 10 min at 4000 rpm
Preparation of columns: addition of 500 μL Preparation solution, centrifugation for 1 min at 12 000 rcf, discard flow-through
Give supernatant of centrifuged samples onto columns, spin for 1 min at 12 000 rcf, discard flow-through
Addition of 500 μL Optional Wash Solution, spin for 1 min at 12 000 rcf, discard flow-through
Addition of 750 μL Wash Solution, spin for 1 min at 12 000 rcf, discard flow-through
Dry columns by centrifuging them for 2 min at 12 000 rcf, subsequently place columns in new collection tubes
Elute DNA with 100 μL Elution solution
According to Sigma-Aldrich Plasmid Miniprep Kit protocol
Construction of the regulator plasmids
Thursday, July 5th
Construction of the lasR regulator plasmid (piG0040)
Competent cells were transformed with piG0028 (K553003, lasR) and selected on chloramphenicol-LB-plates. Plasmid DNA was extracted by performing a miniprep. The relevant plasmid sequence was sequenced by Microsynth using the primers oiG0001 and oiG0002. Amplification of lasR by PCR using oiG0003 and oiG0004 resulted in the fragment fiG0004 (1.0 kb). The vector piG0034 (pSEVA 181) was digested with the restriction enzymes HindIII and PacI to fiG0001 (3.0 kb). The two fragments, fiG0001 and fiG0004, were assembled using Gibbson assembly. Thus we used the plasmid backbone of piG0034 and lasR of piG0028 to construct the lasR regulator plasmid piG0040. Competent cells were transformed with piG0040 and selected on ampicillin-LB-plates. Colony PCR using the primers oiG0035 and oiG0036 was conducted to check the size of the inserted fragment (expected bands at 1.0 and 1.2 kb). The sequence was verified by Microsynth using the same primers.



Construction of the luxR regulator plasmid (piG0041)
Competent cells were transformed with piG0008 (F2620, luxR) and selected on chloramphenicol-LB-plates. Plasmid DNA was extracted by performing a miniprep. The relevant plasmid sequence was sequenced by Microsynth using the primers oiG0001 and oiG0002. Amplification of luxR by PCR using oiG0005 and oiG0006 resulted in the fragment fiG0005 (0.8 kb). The backbone of the vector piG0040 (lasR in pSEVA 181) was amplified by PCR using oiG0007 and oiG0008. This fragment, fiG0011 (3.2 kb), and fiG0005 were assembled using Gibbson assembly. Thus we formally replaced lasR by luxR to construct the luxR regulator plasmid piG0041. Competent cells were transformed with piG0041 and selected on ampicillin-LB-plates. Colony PCR using the primers oiG0035 and oiG0036 was conducted to check the size of the inserted fragment (expected bands at 1.1 and 1.2 kb). The sequence was verified by Microsynth using the same primers.


Construction of the rhlR regulator plasmid (piG0042)
Competent cells were transformed with piG0023 (C0171, rhlR) and selected on chloramphenicol-LB-plates. Plasmid DNA was extracted by performing a miniprep. The relevant plasmid sequence was sequenced by Microsynth using the primers oiG0001 and oiG0002. Amplification of rhlR by PCR using oiG0009 and oiG0010 resulted in the fragment fiG0006 (0.8 kb). The backbone of the vector piG0040 (lasR in pSEVA 181) was amplified by PCR using oiG0011 and oiG0012. This fragment, fiG0012 (3.2 kb), and fiG0006 were assembled using Gibbson assembly. Thus we formally replaced lasR by rhlR to construct the rhlR regulator plasmid piG0042. Competent cells were transformed with piG0042 and selected on ampicillin-LB-plates. Colony PCR using the primers oiG0035 and oiG0036 was conducted to check the size of the inserted fragment (expected bands at 1.1 and 1.2 kb). The sequence was verified by Microsynth using the same primers.


Construction of the luxR regulator plasmids with alternative constitutive promoters (piG0046 and piG0047)
The promoter site of the luxR regulator plasmid piG0041 was mutated by QuikChange site-specific mutagenesis to produce promoters of different strength. The primers oiG0031 and oiG0032 were used to establish piG0047, a plasmid with a promoter of intermediate strength (J23111). Analoguos the primers oiG0033 and oiG0034 were used to construct piG0046, a plasmid with a weak promoter (J23109). Competent cells were transformed with piG0046 and piG0047 and selected on ampicillin-LB-plates. The sequence was verified by Microsynth using the primers oig0035 and oiG0036.
Preparation of DNA from iGEM kit
Friday, July 4th
E coli transformation
Addition of 10 μL H2O to appropriate well, wait for 5 min, transfer into sterile tube
Transformation of E. coli with pSEVA281 A-C2 (sfGFP), pSEVA271 C-A9 (mCherry) and piG0030:
Addition of 1 μL DNA to 75 μL competent cells (thawed on ice), put samples on ice for approximately 20 min
Heat shock: 90 s at 42 °C
Addition of SOC to samples, let cells recover for ca. 1h at 37 °C, 220 rpm
Plate 100 μL of bacteria suspension on LB-agar-plates containing the appropriate antibiotics (pSEVA281 A-C2, pSEVA271 C-A9: Kanamycin (50 μg/L), piG0030: Chloramphenicol (34 μg/L))
Let bacteria grow overnight at 37 °C
Sequencing, Transformation, antibiotic stock preparation
Thursday, July 3rd
Sequencing results
Biobricks of the plasmids piG0001, piG0015 and piG002 show the correct sequences
E coli Transformation
Transformation of E. coli with pSEVA181, pSEVA271, pSEVA291, pSEVA561.
Addition of 1 μL DNA to 75 μL competent cells (thawed on ice), put samples on ice for approximately 20 min
Heat shock: 90 s at 42 °C
Addition of SOC to samples, let cells recover for ca. 1h at 37 °C, 220 rpm
Plate 100 μL of bacteria suspension on LB-agar-plates containing the appropriate antibiotics (pSEVA181: Ampicillin (200 μg/L), pSEVA271 and pSEVA291: Kanamycin (50 μg/L), pSEVA561: Tetracycline (20 μg/L))
Let bacteria grow overnight at 37 °C
Antibiotic stock preparation
Preparation of antibiotics stock:
2 g Ampicillin were dissolved in 10 mL H2O and sterile filtered
0.5 g Kanamycin were dissolved in 10 mL H2O and sterile filtered
0.34 g Chloramphenicol were dissolved in 10 mL Ethanol
Week 6: Project planning, Gibson assemblies planning, First Matlab simulations
Wednesday, July 2nd
- We have a clear organization of the different experiments we would like to perform.


We want to optimize quorum sensing to have non-leaky non-cross-talking constructs. We want to prevent cross-talk between integrases and check their dynamics with different quorum sensing promoters. Then we will test the XOR gate without production of LuxI, to check how it works without the loop, and we will finally test our final construct with the loop, hoping that the delay between GFP production and the possible second switching due to AHL production is long enough to have enough fluorescence.
The red line is our critical timeline. Numbers of days are very optimistic,therefore we multiply the whole timeline by 2. The stars stand for modeling inputs.

- We have also clear plans for the Gibson assemblies to perform. As an example, here is the plan for regulator plasmids :

- We have a facebook page !
- We simulated quorum sensing without leakiness on Matlab, with parameters from the literature.
Team:ETH Zurich/labblog/20140701
Team:ETH Zurich/labblog/20140630
Team:ETH Zurich/labblog/20140629
Team:ETH Zurich/labblog/20140628
Team:ETH Zurich/labblog/20140627
Construction of the regulator plasmids
Thursday, July 5th
Construction of the lasR regulator plasmid (piG0040)
Competent cells were transformed with piG0028 (K553003, lasR) and selected on chloramphenicol-LB-plates. Plasmid DNA was extracted by performing a miniprep. The relevant plasmid sequence was sequenced by Microsynth using the primers oiG0001 and oiG0002. Amplification of lasR by PCR using oiG0003 and oiG0004 resulted in the fragment fiG0004 (1.0 kb). The vector piG0034 (pSEVA 181) was digested with the restriction enzymes HindIII and PacI to fiG0001 (3.0 kb). The two fragments, fiG0001 and fiG0004, were assembled using Gibbson assembly. Thus we used the plasmid backbone of piG0034 and lasR of piG0028 to construct the lasR regulator plasmid piG0040. Competent cells were transformed with piG0040 and selected on ampicillin-LB-plates. Colony PCR using the primers oiG0035 and oiG0036 was conducted to check the size of the inserted fragment (expected bands at 1.0 and 1.2 kb). The sequence was verified by Microsynth using the same primers.
Construction of the luxR regulator plasmid (piG0041)
Competent cells were transformed with piG0008 (F2620, luxR) and selected on chloramphenicol-LB-plates. Plasmid DNA was extracted by performing a miniprep. The relevant plasmid sequence was sequenced by Microsynth using the primers oiG0001 and oiG0002. Amplification of luxR by PCR using oiG0005 and oiG0006 resulted in the fragment fiG0005 (0.8 kb). The backbone of the vector piG0040 (lasR in pSEVA 181) was amplified by PCR using oiG0007 and oiG0008. This fragment, fiG0011 (3.2 kb), and fiG0005 were assembled using Gibbson assembly. Thus we formally replaced lasR by luxR to construct the luxR regulator plasmid piG0041. Competent cells were transformed with piG0041 and selected on ampicillin-LB-plates. Colony PCR using the primers oiG0035 and oiG0036 was conducted to check the size of the inserted fragment (expected bands at 1.1 and 1.2 kb). The sequence was verified by Microsynth using the same primers.
Construction of the rhlR regulator plasmid (piG0042)
Competent cells were transformed with piG0023 (C0171, rhlR) and selected on chloramphenicol-LB-plates. Plasmid DNA was extracted by performing a miniprep. The relevant plasmid sequence was sequenced by Microsynth using the primers oiG0001 and oiG0002. Amplification of rhlR by PCR using oiG0009 and oiG0010 resulted in the fragment fiG0006 (0.8 kb). The backbone of the vector piG0040 (lasR in pSEVA 181) was amplified by PCR using oiG0011 and oiG0012. This fragment, fiG0012 (3.2 kb), and fiG0006 were assembled using Gibbson assembly. Thus we formally replaced lasR by rhlR to construct the rhlR regulator plasmid piG0042. Competent cells were transformed with piG0042 and selected on ampicillin-LB-plates. Colony PCR using the primers oiG0035 and oiG0036 was conducted to check the size of the inserted fragment (expected bands at 1.1 and 1.2 kb). The sequence was verified by Microsynth using the same primers.
Construction of the luxR regulator plasmids with alternative constitutive promoters (piG0046 and piG0047)
The promoter site of the luxR regulator plasmid piG0041 was mutated by QuikChange site-specific mutagenesis to produce promoters of different strength. The primers oiG0031 and oiG0032 were used to establish piG0047, a plasmid with a promoter of intermediate strength (J23111). Analoguos the primers oiG0033 and oiG0034 were used to construct piG0046, a plasmid with a weak promoter (J23109). Competent cells were transformed with piG0046 and piG0047 and selected on ampicillin-LB-plates. The sequence was verified by Microsynth using the primers oig0035 and oiG0036.
Week 5: Assembly strategy and steady states derivation
Wednesday, June 25th
- We will use Gibson assembly to assemble our constructs, and have now a construct assembly strategy. We know the list of experiments we want to carry (cross-talk between quorum sensing molecules, quorum sensing leakiness with or without riboswitch, cross-talk between integrases, integrase activity and logic gate behaviour) and the exact sequences of base units we will assemble (lux, rhl, las promoter and repressor units, reporter units, logic units with 3 different integrases, constitutive AHL production units for diffusion test, backbone units).

- From the modeling side, we calculated analytically the steady state of the integrase and terminator modules, and it appears that the deterministic model gives a binary steady state corresponding to an XOR gate. This is what we expected, it confirms that deterministic model in our case is interesting for the study of dynamics of the whole system, and not for steady state calculations. So we are now moving to dynamic studies.
Team:ETH Zurich/labblog/20140624
Team:ETH Zurich/labblog/20140623
Team:ETH Zurich/labblog/20140622
Team:ETH Zurich/labblog/20140621
Team:ETH Zurich/labblog/20140620
Team:ETH Zurich/labblog/20140619
Week 4: Improving plasmid design, Modularization of the whole model
Wednesday, June 18th
- We improved a little bit the plasmid design, determined which parts will be taken from the iGEM registry, and which parts we have to synthesize.

Sensor plasmids will be approx. 9.3kb long, and logic plasmids will be approx. 5,2kb long.
- From the modeling side, the whole model has been modularized and we have calculated and simulated steady state for the quorum sensing module.
- GeneScript, Microsynth, EraSynbio are willing to support us !
Team:ETH Zurich/labblog/20140617
Team:ETH Zurich/labblog/20140616
Team:ETH Zurich/labblog/20140615
Team:ETH Zurich/labblog/20140614
Team:ETH Zurich/labblog/20140613
Team:ETH Zurich/labblog/20140612
Week 3: Plasmid design started, Modeling started
Wednesday, June 11th
- We started plasmid design :

ΦC31 and Bxb1 are integrases. LuxR an RhlR are quorum sensing repressors. A riboswitch construct is placed around quorum sensing constructs to prevent leakiness of lux and rhl promoters. Type p colonies produce AHL by expressing the enzyme LuxI. Type q colonies produce Rhl by expressing the enzyme RhlI. In fact, we don't know yet which quorum sensing systems we will use. We will have to perform cross-talk experimetns in order to choose the ones that are the most orthogonal.
- We tried to print our first agar millifluidic chip : we printed it too small, and the printer had resolution problems.
- We wrote all reactions and found parameters from the literature for our model.
Team:ETH Zurich/labblog/20140610
Team:ETH Zurich/labblog/20140609
Team:ETH Zurich/labblog/20140608
Team:ETH Zurich/labblog/20140607
Team:ETH Zurich/labblog/20140606
Team:ETH Zurich/labblog/20140605
Week 2: Investigating microfluidics, Writing a modeling pipeline
Wednesday, June 4th
This week, we have talked with people from the microfluidics group and it came out that we should be able to use beads to encapsulate our cells and use a microfluidic chip. Therefore we will try to develop this possibility in parallel with the 3D-printed agar chip.
We have set up a modeling pipeline. We divided the modeling project into 3 parts :
- diffusion,
- parameter fitting,
- modeling of the genetic circuit itself, which comprises
- a deterministic model
- a stochastic model.
We know that the modeling challenges will be to model how integrases work, and to model the delay between reception of a quorum sensing signal and production of a quorum sensing signal by the receiver cell. Indeed, a colony could switch itself OFF when it should be ON, if it receives QS1 and produces QS2, for example. If a delay is present between reception of QS1 and production of QS2, the colony will produce GFP before it is switched OFF, and as GFP is stable enough, it will stay visible during a long time and the self-switching OFF won't be observed. This delay should be modeled.
Team:ETH Zurich/labblog/20140603
Team:ETH Zurich/labblog/20140602
Team:ETH Zurich/labblog/20140601
Team:ETH Zurich/labblog/20140530
Team:ETH Zurich/labblog/20140529
Week 1 : Project selected
Wednesday, May 28th
After more than one month of endless meetings and passionate debates, we finally chose the project that will keep us occupied in the next five months. From the beautiful pattern made by the Sierpinski triangles, we will focus on cellular automata and try to implement one.
Sierpinski triangles appear when the rule 90 is followed by every cell on the grid :

Ideally we will use a microfluidic chip. We could also use a 3D printed agar plate like this one to load the colonies. On this grid we can implement the rule 6, which is the simplification of rule 90 considered as a rule with 2 inputs : each cell computes a simple XOR gate of its two parents.


The logic part will be built with integrases and the colony-to-colony communication will use quorum sensing.
Every colony will receive two quorum sensing signals (QSp and QSq) from the two cells above it. These two signals trigger the production of two different integrases r and s in the colony. Integrases enable to build biological XOR logic gates by switching twice a terminator. Indeed, every integrase can switch the terminator only once. Thus if the colony produces only r or only s, the terminator is switched only once, so the terminator is OFF, and GFP and QS1 or QS2 are produced (depending on the colony). If the colony produces r and s, the terminator is switched twice, so it is ON and it blocks expression of GFP and of the quorum sensing molecule.

We need to :
- find orthogonal quorum sensing molecules and orthogonal integrases
- discuss with microfluidics experts to check if using microfluidics is possible and presents advantages in our case
- find possible parts in the registry for integrases, and design plasmids
Team:ETH Zurich/labblog/20140527 Team:ETH Zurich/labblog/20140526 Team:ETH Zurich/labblog/20140525 Team:ETH Zurich/labblog/20140524 Team:ETH Zurich/labblog/20140523 Team:ETH Zurich/labblog/20140522 Team:ETH Zurich/labblog/20140521 Team:ETH Zurich/labblog/20140520 Team:ETH Zurich/labblog/20140519 Team:ETH Zurich/labblog/20140518 Team:ETH Zurich/labblog/20140517 Team:ETH Zurich/labblog/20140516 Team:ETH Zurich/labblog/20140515 Team:ETH Zurich/labblog/20140514 Team:ETH Zurich/labblog/20140513 Team:ETH Zurich/labblog/20140512 Team:ETH Zurich/labblog/20140511
Team:ETH Zurich/labblog/20140509 Team:ETH Zurich/labblog/20140508 Team:ETH Zurich/labblog/20140507 Team:ETH Zurich/labblog/20140506 Team:ETH Zurich/labblog/20140505 Team:ETH Zurich/labblog/20140504 Team:ETH Zurich/labblog/20140503 Team:ETH Zurich/labblog/20140502 Team:ETH Zurich/labblog/20140501